Fármaco
 | A introdución deste artigo precisa dunha ampliación, redución, carece de contexto ou non fornece un resumo axeitado do artigo segundo indica o libro de estilo da Galipedia. Pode axudar a mellorar este artigo e outros en condicións semellantes. |
 | Advertencia: A Wikipedia non dá consellos médicos. Se cre que pode requirir tratamento, por favor, consúltello ao médico. |

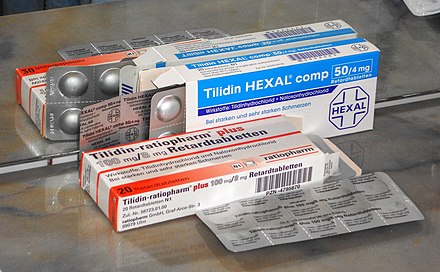
Un fármaco[1] (gr. φάρμακον), ás veces referido como medicina, menciña ou medicamento[2] é toda substancia química purificada empregada na prevención, diagnóstico, tratamento, mitigación ou cura dunha doenza,[3][4] para evitar a aparición dun proceso fisiolóxico non desexado ou ben para modificar condicións fisiolóxicas con fins específicos. É unha substancia, simple ou composta, utilizada como finalidade terapéutica contra as manifestacións patolóxicas, tanto se se produce a curación - suprimindo a causa da enfermidade - coma se só atenúa os síntomas.[5]
Historia
[editar | editar a fonte]- Artigo principal: Historia da farmacia.
Desde as máis antigas civilizacións o home utilizou como forma de alcanzar melloría en distintas enfermidades produtos de orixe vexetal, mineral, animal ou nos últimos tempos sintéticos.[6] O coidado da saúde estaba en mans de persoas que exercían a dobre función de médicos e farmacéuticos, estes eran en realidade médicos que preparan os seus propios remedios curativos, chegando algún deles a alcanzar un grande renome na súa época, como é o caso do grego Galeno (130-200). Del provén o nome da Galénica, como a forma adecuada de preparar, dosificar e administrar os fármacos. Na cultura romana existían numerosas formas de administrar as substancias utilizadas para curar enfermidades. Así, utilizábanse os electuarios que eran unha mestura de varios pos de herbas e raíces medicinais aos que se lles engadía unha porción de mel fresco. O mel ademais de ser a substancia que servía como vehículo dos principios activos, daba mellor sabor ao preparado. En ocasións usábase azucre. Tamén adoitaba utilizarse un xarope o cal xa contiña azucre, no canto de auga, disolto e o conxunto preparábase formando unha masa pastosa.[7] Precisamente Galeno fixo famosa a gran triaga á que dedicou unha obra completa, e que consistía nun electuario que chegaba a conter máis de 60 principios activos diferentes.[8] Pola importancia de Galeno na Idade Media, a triaga fíxose moi popular durante esta época, deixando de estar autorizada para o seu uso en España en pleno século XX.[9] É precisamente na Idade Media onde comeza a súa actividade o farmacéutico separado do médico. Na súa botica realiza as súas preparacións maxistrais, entendidas como a preparación individualizada para cada paciente dos remedios prescritos, e agrúpanse en gremios xunto aos médicos. No renacemento vaise producindo unha separación máis clara da actividade farmacéutica fronte a dos médicos, cirurxiáns e especieiros, mentres que se vai producindo unha revolución no coñecemento farmacéutico que se consolida como ciencia na Idade Moderna. A formulación maxistral é a base da actividade farmacéutica conxuntamente coa formulación oficinal, debido ao nacemento e proliferación de farmacopeas e formularios, e esta situación continúa ata a segunda metade do século XIX.[10]
A partir deste momento empezan a aparecer os específicos, que consistían en medicamentos preparados industrialmente por laboratorios farmacéuticos. É así, que as formas galénicas non adquirirán verdadeiro protagonismo ata ao redor de 1940, cando a industria farmacéutica desenvólvese e estas comezan a fabricarse en grandes cantidades. Desde entón ata hoxe en día os xeitos en que se presentan os medicamentos foron evolucionando e a diversidade que atopamos no mercado é moi ampla.[11]
Forma farmacéutica
[editar | editar a fonte]Forma galénica ou forma farmacéutica é a disposición individualizada a que se adaptan os fármacos (principios activos) e excipientes (materia farmacoloxicamente inactiva) para constituír un medicamento.[12] Ou dito doutra forma, a disposición externa que se dá ás substancias medicamentosas para facilitar a súa administración.
O primeiro obxectivo das formas galénicas é normalizar a dose dun medicamento, por iso tamén se coñecen como unidades posolóxicas. Ao principio elaboráronse para poder establecer unidades que tivesen unha dose fixa dun fármaco co que se puidese tratar unha determinada patoloxía".[11]
A importancia da forma farmacéutica reside en que determina a eficacia do medicamento, xa sexa liberando o principio activo de xeito lento, ou no seu lugar de maior eficiencia no tecido branco, evitar danos ao paciente por interacción química, solubilizar substancias insolubles, mellorar sabores, mellorar aspecto etc.
Clasificación
[editar | editar a fonte]Os medicamentos divídense en cinco grupos:
- Especialidade farmacéutica: É o medicamento de composición e información definidas, de forma farmacéutica e dosificación determinadas, preparado para o seu uso medicinal inmediato, disposto e acondicionado para a súa dispensación ao público, con denominación, embalaxe, envase e etiquetado uniformes segundo o dispoñan as autoridades sanitarias.
- Fórmula maxistral: É o medicamento destinado a un paciente individualizado, preparado polo farmacéutico, ou baixo a súa dirección, para cumprimentar expresamente unha prescrición facultativa detallada das substancias medicinais que inclúe, segundo as normas técnicas e científicas da arte farmacéutica, dispensado na súa farmacia ou servizo farmacéutico e coa debida información ao usuario.
- Preparado ou fórmula oficinal: É aquel medicamento elaborado e garantido por un farmacéutico ou baixo a súa dirección, dispensado na súa oficina de farmacia ou servizo farmacéutico, enumerado e descrito polo Formulario, destinado á entrega directa aos enfermos aos que abastece dita farmacia ou servizo farmacéutico.
- Medicamento prefabricado: É o medicamento que non se axusta á definición de especialidade farmacéutica e que se comercializa nunha forma farmacéutica que pode utilizarse sen necesidade de tratamento industrial e ao que a autoridade farmacéutica outorgue autorización e inscriba no rexistro correspondente.
- Medicamento en investigación: Forma farmacéutica dunha substancia activa ou placebo, que se investiga ou se utiliza como referencia nun ensaio clínico, incluídos os produtos con autorización de comercialización cando se utilicen ou combinen, na formulación ou no envase, de forma diferente á autorizada, ou cando se utilicen para tratar unha indicación non autorizada, ou para obter máis información sobre un uso autorizado.
Ademais, poden recibir algúns cualificativos específicos como:
- Medicamento citostático
- Medicamento compasivo
- Medicamento esencial
- Medicamento heroico: de acción moi enérxica que só se aplica en casos extremos.[13]
- Medicamento orfo
- Medicamento milagroso
- Medicamentos similares
Segundo a prescrición médica
[editar | editar a fonte]En España e algúns países latinoamericanos, os medicamentos dispénsanse, distribúen ou venden exclusivamente nas farmacias. Existen dous tipos de medicamentos segundo a prescrición médica:
- Medicamento de venda libre: Son aqueles medicamentos que se distribúen libremente nas farmacias, sen necesidade de prescrición ou receita médica. Divídense en dúas categorías:
- As especialidades farmacolóxicas publicitarias (EFP) correspóndense con medicamentos publicitados nos medios de comunicación de masas como, por exemplo, a televisión.[Cómpre referencia]
- Os produtos OTC ("Over the Counter") son fármacos destinados ao alivio, tratamento ou prevención de afeccións menores, cos que se posúe unha ampla experiencia de uso e foron expresamente autorizados como tales.
- Medicamento con receita médica: Son aqueles medicamentos receitados por un médico para o tratamento dunha enfermidade ou síntoma en concreto.
Segundo o dereito de explotación
[editar | editar a fonte]- Medicamento con patente: son aqueles medicamentos de investigación propia do laboratorio que os comercializa, suxeitos á protección comercial que brindan as axencias internacionais de patentes.
A patente non se limita á molécula, senón tamén á formulación, mecanismo de produción, ou asociación con outras moléculas. Mediante sucesión de patentes as casas farmacéuticas conseguen prolongar o período de exclusividade das súas presentacións comerciais, aínda cando presentacións anteriores da mesma molécula queden libres.
- Medicamento xenérico: aquelas presentacións de moléculas que xa non están protexidas pola patente do seu investigador.
Poden ser libremente producidas por outros laboratorios e adoitan supoñer un menor prezo. As distintas Axencias do medicamento e organizacións reguladores nacionais aseguran as similares bioequivalencia e biodispoñibilidade dos medicamentos xenéricos fronte a aqueles que lles son referencia.
Segundo a vía de administración
[editar | editar a fonte]- Artigo principal: Vías de administración de fármacos.
Existen numerosas formas de clasificar as formas galénicas, segundo o factor que teñamos en conta: o seu estado físico, a vía de administración, a orixe dos seus compoñentes etcétera. Non obstante a máis utilizada e a máis útil desde o punto de vista da medicina é a clasificación segundo a vía de administración que usen.
Oral
[editar | editar a fonte]A maior parte dos fármacos administrados vía oral buscan unha acción sistémica, tras un proceso previo de absorción entérica. Na absorción oral interveñen factores dependentes do individuo e outros dependentes dos fármacos que van influír na maior ou menor eficacia do fármaco administrado. Así mesmo, a vía oral é motivo frecuente de interaccións farmacolóxicas.
A vía oral constitúe a vía máis utilizada de administración de fármacos, subdividindose á súa vez, en formas líquidas e formas sólidas.
As formas orais líquidas. Non suscitan problemas de disgregación ou de disolución no aparello dixestivo, o que condiciona unha acción terapéutica máis rápida. Pola contra non están protexidas, en caso de reactividade, fronte aos mollos dixestivos. Resultan de elección particularmente en nenos. Os líquidos para administración oral son habitualmente solucións, emulsións ou suspensións que conteñen un ou máis principios activos disoltos nun vehículo apropiado. Os vehículos poden ser:
- Acuosos: serven para disolver principios activos hidrosolubles. Os máis comúns son os xaropes (que conteñen unha alta concentración de azucre, ata un 64% en peso).
- Mucilaxes: líquidos viscosos resultantes da dispersión de substancias gomosas (goma arábiga, tragacanto, agar, metilcelulosa) en auga. Úsanse, sobre todo, para preparar suspensións e emulsións.
- Hidroalcohólicos: os elixires son solucións hidroalcohólicas (25% alcohol) edulcoradas utilizadas para disolver substancias solubles en auga e alcohol.
Estas formas líquidas poden conter tamén substancias auxiliares para a conservación, estabilidade ou o enmascaramento do sabor do preparado farmacéutico (conservantes, antimicrobianos, antioxidantes, tampóns, solubilizantes, estabilizantes, aromatizantes, edulcorantes e colorantes autorizados).
As formas farmacéuticas líquidas para administración oral máis usuais son:
Gotas
[editar | editar a fonte]Son solucións nas que o principio activo está concentrado.
Xaropes
[editar | editar a fonte]- Artigo principal: Xarope.
Forma farmacéutica que consiste nunha solución acuosa con alta concentración de carbohidratos tales como sacarosa, sorbitol, dextrosa etc.; de consistencia viscosa, na que se atopa disolto o ou os principios activos e aditivos.[14]
Tisanas
[editar | editar a fonte]As tisanas son infusións con baixa concentración de principios activos. Son as máis seguras durante a súa utilización, a base é unha ou varias herbas medicinais e dilúense xeralmente en auga quente; poden ser endulzadas con mel para mellorar o seu sabor. Existen diferentes preparacións:
- Infusións:
Extráense as propiedades das plantas vertendo auga quente sobre ela e deixando repousar durante 5 ou 10 minutos.
- Cocementos:
Férvese a parte máis dura da planta durante uns 30 minutos a fogo lento.
- Maceracións:
Déixase repousar a herba medicinal desde unhas 6 horas ata semanas para extraer o principio activo.
Elixires
[editar | editar a fonte]
Os elixires son unha forma farmacéutica que consiste nunha solución hidroalcohólica, que contén o/ou os principios activos e aditivos; contén xeralmente substancias saborizantes, así como aromatizantes. O contido de alcohol pode ser do 5 ao 18 por cento..[14]
Suspensións
[editar | editar a fonte]As suspensións son mesturas heteroxéneas formadas por un sólido en po (soluto) e/ou en pequenas partículas non solubles (fase dispersa) que se dispersan nun medio líquido (dispersante ou dispersora). Sólido en líquido que non é soluble neste.
Suspensión extemporánea
[editar | editar a fonte]Aquela que, pola súa pouca estabilidade, prepárase no momento de ser administrada.
Viales bebibles
[editar | editar a fonte]
Formas orais sólidas. As formas sólidas, presentan unha maior estabilidade química debido á ausencia de auga, o que lles confire tempos de reposición máis longos. Ademais, estas formas galénicas permiten resolver posibles problemas de incompatibilidades, enmascarar sabores desagradables e ata regular a liberación dos principios activos.
As formas farmacéuticas sólidas máis frecuentes para administración oral son:
- Comprimidos
Os comprimidos son formas farmacéuticas sólidas que conteñen, en cada unidade, un ou varios principios activos. Obtéñense aglomerando, por compresión, un volume constante de partículas. Adminístranse xeralmente por deglutición, aínda que se poden dar outras posibilidades.

- Cápsulas
As cápsulas son preparacións de consistencia sólida formadas por un receptáculo duro ou brando, de forma e capacidade variable, que conteñen unha unidade posológica de medicamento. Este contido pode ser de consistencia sólida, líquida ou pastosa e estar constituído por un ou máis principios activos, acompañados ou nó de excipientes. O receptáculo desfarase pola acción dos mollos gástricos ou entéricos, segundo a formulación, liberando entón o principio activo.
- Granulados
Os granulados son agregados de partículas de pos que inclúen principios activos, azucres e coadxuvantes diversos. Preséntanse en forma de pequenos grans de grosor uniforme, forma irregular e máis ou menos porosidade. Existen granulados de distintos tipos: efervescentes, recubertos, gastrorresistentes e de liberación modificada.
- Selos
Os selos son cápsulas cun receptáculo de amidón. Practicamente, foron desprazados polas cápsulas duras.
- Pílulas
As pílulas son preparacións sólidas e esféricas, destinadas a ser deglutidas integramente. Cada unidade contén un ou máis principios activos interpostos nunha masa plástica. Atópanse en franco desuso, sendo desprazadas polos comprimidos e cápsulas.
- Pastillas
As pastillas son para disolver na cavidade bucal. Diferéncianse das pílulas polo tamaño e dos comprimidos pola técnica de elaboración. Os seus constituíntes principais son a sacarosa, un aglutinante e un ou máis principios activos.
- Pastillas oficinais ou trociscos
As pastillas oficinais ou trociscos presentan unha consistencia semisólida e están constituídas primordialmente polos principios activos e goma arábiga como aglutinante. Adoitan recubrirse, para a súa mellor conservación, con parafina ou azucre en po (xeado). Empréganse para a vehiculización de antitusíxenos e antisépticos pulmonares.
- Liofilizados
Os liofilizados son preparacións farmacéuticas que se acondicionan en forma de dose unitarias e se liofilizan a continuación. Son formas moi porosas e hidrófilas e facilmente dispersables en auga.
- Colutorios
Os colutorios son solucións acuosas viscosas para o tratamento tópico de afeccións bucais e que aplícanse cun pincel ou unha espátula. Estritamente un colutorio diferénciase dunha enxaugadura bucal ao non levar viscosizantes e pode ser sólido (pos ou comprimidos) para diluír antes de usar.[15]
Sublingual
[editar | editar a fonte]Unha forma especial de administración oral é a vía sublingual. Nesta vía normalmente, utilízanse comprimidos que se disolven debaixo da lingua absorbéndose directamente. Ten o inconveniente de ser exclusivamente permeable ao paso de substancias non iónicas, moi liposolubles. Isto fai que só poidan administrarse por esta vía fármacos de gran potencia terapéutica como a nitroglicerina ou o isosorbide. Utilízase para conseguir unha acción terapéutica rápida ou para fármacos que posúan un alto grao de metabolización hepática, se degraden polo mollo gástrico ou non sexan absorbidos por vía oral. No entanto tamén se atopan no mercado presentacións por comodidade do usuario.
Parenteral
[editar | editar a fonte]A biodispoñibilidade dun fármaco administrado vía parenteral depende das súas características fisicoquímicas, da forma farmacéutica e das características anatomofisiolóxicas da zona de inxección:
- Vía intravenosa
Por vía intravenosa proporciona un efecto rápido do fármaco e unha dosificación precisa, sen problemas de biodispoñibilidade. Pode presentar, no entanto, graves inconvenientes, como a aparición de tromboflebite, así como problemas de incompatibilidades entre dous principios activos administrados conxuntamente na mesma vía. Ten o inconveniente de que non permite a administración de preparados oleosos debido á posibilidade de orixinar unha embolia graxa. Tampouco poderán usarse produtos que conteñan compoñentes capaces de precipitar algún compoñente sanguíneo ou hemolizar os hemacias.
- Vía intraarterial
A vía intraarterial é utilizada no tratamento quimioterápico de determinados cancros; permite obter unha máxima concentración do fármaco na zona tumoral, cuns mínimos efectos sistémicos.
- Vía intramuscular
A vía intramuscular utilízase para fármacos non absorbibles por vía oral ou ante a imposibilidade de administración do fármaco ao paciente por outra vía xa que admite o ser utilizada para substancias irritantes. Numerosos factores van influír na biodispoñibilidade do fármaco por vía IM (vascularización da zona de inxección, grao de ionización e liposolubilidade do fármaco, volume de inxección etc.). Esta vía é moi utilizada para a administración de preparados de absorción lenta e prolongada (preparados “depot”).
- Vía subcutánea
A vía subcutánea de características similares á anterior pero ao ser a pel unha zona menos vascularizada, a velocidade de absorción é moito menor. Con todo, dita velocidade pode ser incrementada ou diminuída por distintos medios. Non pode utilizarse para substancias irritantes xa que podería producir necrose do tecido.
Outras vías parenterais
[editar | editar a fonte]De uso menos frecuente son a intradérmica, a intraaracnoidea ou intratecal, epidural, intradural, intraósea, intraarticular, peritoneal ou a intracardiaca.
Os preparados para administración parenteral son formulacións estériles destinadas a ser inxectadas ou implantadas no corpo humano. A continuación enuméranse cinco das máis representativas:
- Preparacións inxectables. Son preparacións do principio activo disolto (disolución), emulsionado (emulsión) ou disperso (dispersión) en auga ou nun líquido non acuoso apropiado.
- Preparacións para diluír previamente á administración parenteral. Solucións concentradas e estériles destinadas a ser inxectadas ou administradas por perfusión tras ser diluídas nun líquido apropiado antes da súa administración.
- Preparacións inxectables para perfusión. Son solucións acuosas ou emulsións de fase externa acuosa, exentas de piróxenos, estériles e, na medida do posible, isotónicas con respecto ao sangue.
- Po para preparacións inxectables extemporáneas. substancias sólidas estériles, dosificadas e acondicionadas en recipientes definidos que, rapidamente tras axitación, en presenza dun volume prescrito de líquido estéril apropiado, dan lugar a solucións practicamente límpidas, exentas de partículas, ou ben a suspensións uniformes.
- Implantes ou pellet. Pequenos comprimidos estériles de forma e tamaño adecuados que garanten a liberación do principio activo ao longo dun tempo prolongado.
Por vía parenteral é posible a liberación retardada ou prolongada dos principios activos a partir do punto de inxección. Isto pódese conseguir realizando diversas manipulacións galénicas. Unha delas consiste en substituír unha solución acuosa por unha oleosa, no caso de que o principio activo sexa liposoluble. O método máis clásico consiste en inxectar derivados pouco hidrosolubles do principio activo, en forma de suspensións amorfas ou cristalinas (p. e., preparacións retard de insulina). Ás veces, o principio activo pode tamén absorberse sobre un soporte inerte desde o que será liberado, ou ben fixarse en forma de microcápsulas, ou incorporarse en liposomas para vectorizar algúns fármacos e, incluso ser tratado quimicamente (profármaco) a fin de modificar as súas propiedades fisicoquímicas.
Rectal
[editar | editar a fonte]Supositorios
[editar | editar a fonte]
Os supositorio son preparados de consistencia sólida de forma cónica e redondeada nun extremo. Teñen unha lonxitude de 3–4 cm e un peso de entre 1-3 g. Cada unidade inclúe un ou varios principios activos, incorporados nun excipiente que non debe ser irritante, o cal debe ter un punto de fusión inferior a 37 °C. Os excipientes dos supositorios poden ser:
- Liposolubles: Son os máis utilizados; entre eles atópanse a manteiga de cacao, os glicéridos semisintéticos e os aceites polioxietilenados saturados.
- Hidrosolubles: polietilenglicois (PEG). Hai moitas outras substancias que se inclúen neles.[16] Hai dous tipos de supositorios, o primeiro deles é o supositorio de glicerogelatina e o segundo deles é o Polietilenglicol e os supositorios de PEG. Os supositorios PEG son bos para os efectos a longo prazo do sistema, pero os supositorios de glicerogelatina son para os efectos locais e rápidos, xa que se disolven rapidamente.
Cápsulas rectais
[editar | editar a fonte]
Solucións e dispersións rectais
[editar | editar a fonte]- Enemas: Son formas galénicas líquidas, de composición variable, destinadas a ser administradas por vía rectal, empregando para iso dispositivos especiais. Poden ter como obxectivo a vehiculización dun principio activo (enemas medicamentosos), o baleirado da ampola rectal (enemas evacuantes) ou administrar unha substancia radio-opaca para a realización de estudos radiolóxicos (enemas opacos).
Pomadas rectais
[editar | editar a fonte]A vía rectal pode utilizarse para conseguir efectos locais ou sistémicos. Neste último caso, só se debe considerar como unha alternativa á vía oral cando esta non poida utilizarse, xa que a absorción polo recto é irregular, incompleta e ademais moitos fármacos producen irritación da mucosa rectal. Un dos poucos exemplos nos que esta forma farmacéutica ten unha indicación preferente é o tratamento das crises convulsivas en nenos pequenos (diacepam).
Tamén existen pomadas rectais para o tratamento das hemorroides externas.
Tópica
[editar | editar a fonte]A vía tópica utiliza a pel e as mucosas para a administración de fármaco. Así pois, isto inclúe mucosa ocular e xenital. A mucosa oral xa foi vista dentro da epígrafe da vía oral.
A característica desta vía é que se busca fundamentalmente o efecto a nivel local, non interesando a absorción dos principios activos. Os excipientes fundamentais para as formulacións galénicas de uso tópico son tres: líquidos, pos e graxas. Estes poden combinarse entre si de numerosas formas para adaptarse ás características do sitio onde se van a aplicar, de aí a variedade de formas galénicas para uso tópico.[17]
| Pasta
(Graxa + po) |
Crema
(Graxa + líquido) | |
| Taboa de tres entradas, coas posibles combinacións entre graxas, líquidos e pos.[17] | ||
Baños
[editar | editar a fonte]Consisten na inmersión de todo ou parte do corpo nunha solución acuosa á que se engaden determinados produtos. Os máis utilizados son os baños coloidais, que tanto mornos como quentes actúan como sedantes e antiprurixinosos. Na súa preparación mestúrase unha cunca de amidón nun litro de auga e posteriormente esta preparación votase á auga do baño. Nos baños oleosos substitúese o amidón por aceites facilmente dispersables, producindo unha suspensión homoxénea. Actualmente utilízanse sobre todo os baños de sales, potenciados polo mundo da cosmética.

Tamén coñecidas en dermatoloxía como curas húmidas abertas son moi útiles en inflamacións superficiais como o eccema. Colócanse comprimidas empapadas en certos líquidos nas zonas secretantes e cámbianse cada 5-10 minutos, ou ben se engade de forma periódica líquido, de tal xeito que se evite o desecamento por efecto da calor da pel. Isto adóitase manter durante 2 horas tres veces ao día. O obxectivo é o arrefriamento que consegue a evaporación do líquido, que actúa como antiinflamatorio. Ademais teñen propiedades antipruriginosas e vasoconstritoras. Xamais irritan e non teñen riscos no seu uso. Os líquidos utilizados poden ser o soro fisiolóxico, etanol ao 10%, o permanganato potásico ao 0.01% ou a solución de Burow, composta por auga, subacetato de chumbo e sulfato alumínico potásico. Unha loción clásica da farmacopea española sería o Agua de alibour, composta por unha solución hidroalcohólica ao 1% e sulfato de cobre, sulfato de cinc e alcanfor e indicada no tratamento do impetixe, dermatite sobreinfectada e dermatose exudativa.[18]
Toques ou pincelacións
[editar | editar a fonte]Adóitanse utilizar para descostrar feridas ou lesións dérmicas. Utilízase o etanol ao 10% ao que se lle pode engadir glicerina ao 5%. O etanol pode causar queimazón, polo que se desaconsella n rexión genital e cara. Unha forma especial serían as pincelacións secantes da farmacopea alemá, que son en realidade locións axitables, constituídas a partes iguais en peso de pos e líquido, aínda que son algo máis cosméticas cunha proporción algo inferior de pos. A estas locións pódeselles engadir principios activos en forma de líquido ou de po. As indicacións son as mesmas que as dos pos (ver despois), xa que en realidade o que se busca é o depósito destes sobre a pel ao evaporarse o líquido da formulación.[17]
Tinturas
[editar | editar a fonte]Son preparacións líquidas, coloreadas (de onde o seu nome), resultado da mestura de hidroalcoholes con drogas secas a temperatura ambiente. As súas propiedades dependen do fármaco engadido, aínda que en liñas xerais son antisépticas e antiprurixinosas. O máis habitual adoita ser o uso de solucións de etanol, éter ou cloroformo con produtos como o mentol, fenol, fucsina, eosina, violeta de xenzá ou breas. Se se resecan demasiado pódese engadir aceite de rícino ao 1%. Adóitanse administrar unha vez ao día e retíranse os restos ao día seguinte antes da nova aplicación.
Forma farmacéutica que consiste nunha presentación líquida, solución ou emulsión que contén o ou os principios activos e aditivos cuxo vehículo é acuoso, alcohólico ou oleoso, e aplícase exteriormente en friccións
Os pos obtéñense por división sucesiva de produtos sólidos e secos ata partículas homoxéneas de tamaño variable. Trala súa fabricación son pasados por barutos que oscilan entre os 0.1 mm de apertura do enreixado (pos moi finos) ata 1 mm. (pos grosos). Aplicados sobre a pel forman unha capa finísima de propiedades refrescantes, vasoconstritoras, antiinflamatorias e antiprurixinosas. Tamén protexen do rozamento entre superficies e exercen protección mecánica. Os máis famosos son os pos de talco, que en dermatoloxía pódense asociar a outras substancias como o óxido de zinc. Tamén se conta con pos de orixe vexetal (amidón, licopodio), e na industria cosmética utilizáronse amplamente engadindo pigmentos inocuos, do tipo do cinabrio, eosina, bentonita, óxido de ferro ou ictiol para peles escuras. Aínda que se poden fabricar pos directamente coas propias substancias activas, non é isto o máis recomendable, sendo mellor a súa incorporación aos pos inertes citados anteriormente. Así, pódense engadir substancias do tipo das sulfamidas, funxicidas (ciclopirox olamina, miconazol), anhidróticos (cloruro de aluminio) etc. Como inconveniente importante ten o que non se deben de utilizar ante presenza de secrecións, pois a mestura co exsudado ou o pus forma unha especie de cemento que favorece a infección, é irritante e dificulta a cicatrización.[19] É especialmente desaconsexable a estendida utilización de pos asociados a produtos antiprurixinosos na varicela, onde poden deixar cicatrices máis intensas do habitual ou a utilización de pos de boro e ácido salicílico, moi estendidos por parte da industria farmacéutica e de efectos non demostrados.[20]
Están compostas por pos e graxas a partes iguais, aínda que se son demasiado espesas pódese reducir a parte correspondente aos pos, aumentando a cantidade correspondente de graxa. En liñas xerais as pastas son secantes, pero manteñen a pel suave e pregable, protexendoa de traumas mecánicos e permitindo a transpiración. Están indicadas en procesos secos (dermatite crónicas) ou moi pouco secretantes e na prevención das úlceras de decúbito, polo seu efecto protector. Poden incorporar principios activos tanto en forma líquida como sólida. As súas contraindicacións principais son as lesións secretantes, infeccións e rexións pilosas. Aplícanse estendéndoas cunha espátula de madeira e retíranse con aceite de oliva ou vaselina líquida. En lugar de graxas sólidas pódense usar aceites, dando lugar a pastas oleosas. Se a unha pasta se lle engaden líquidos (auga), atopámonos ante a pasta acuosa ou pasta refrescante.
Pomadas ou ungüentos
[editar | editar a fonte]Son graxas ou substancias de parecidas características que presenten aspecto semisólido a 25 °C. É esta propiedade física o que realmente as define xa que a composición química é enormemente variada.[17] O termo pomada é habitualmente usado na literatura hispánica, mentres que o termo ungüento é de uso preferente no ámbito anglosaxón, aínda que por efecto da preponderancia científica, este vaise estendendo como forma principal de uso. Entre non entendidos aplícase o termo pomada a calquera presentación sólida de aplicación na pel, sexa crema, xel ou pomada, da mesma forma que chaman pastilla á maioría das formas galénicas de administración oral. As súas indicacións son amplas: melloran a pel seca e gretada, son excelentes para retirar costras ou escamas e son medianamente toleradas en zonas pilosas. Admiten multitude de principios activos ben en solución, os principios activos liposolubles, ben en forma de po ou en dispersión coloidal para os insolubles en graxas. O seu contraindicación máis importante son os procesos inflamatorios agudos, tanto máis contraindicada canto máis húmido sexa o proceso.

Unha emulsión é unha mestura máis ou menos homoxénea de dous líquidos inmiscíbeis. Un líquido (a fase dispersa) é dispersado noutro (a fase continua ou fase dispersante). Moitas emulsións son emulsións de aceite e auga.
Respecto dos anteriores é de creación moito posterior. Son emulsións semisólidas de polímeros orgánicos (metilcelulosa, agar, xelatina, propilenglicol, galato de propilo, edetato disódico, carboxipolímero) nun líquido (auga).[21] Adóitase engadir hidróxido sódico ou ácido clorhídrico para axustar o pH. Son, pois, bases incoloras, claras, non graxas, miscibles con auga. Pódense considerar como un sistema coloidal onde a fase continua é sólida e a dispersa é líquida. Nesta fase líquida pode incorporar principios activos en solución. A temperatura ambiente son sólidos ou semisólidos, fluidificándose ao ser quentadas. Polo seu aspecto cosmético adóitanse utilizar en zonas pilosas ou estreitas (conduto auditivo externo, fosas nasais). Trala súa aplicación desaparecen rápida e completamente.

Forma galénica utilizada para o coidado do cabelo e do coiro cabeludo.
Forma farmacéutica que consiste nunha solución que contén o ou os principios activos e aditivos, aplicable unicamente á conxuntiva ocular. Debe ser totalmente transparente, estéril, isotónica e cun pH neutro ou próximo á neutralidade.[22]
As gotas óticas e nasais son unha forma galénica para uso tópico consistente nun líquido acuoso ou oleoso no que van incorporados os principios activos e que se utilizan no conduto auditivo externo ou as fosas nasais respectivamente.
As súas propiedades son similares ás dos colirios, salvo a esixencia da esterilidade.[22] Acorde á gran cantidade de principios activos que tolera esta forma galénica, as indicacións tamén son amplas:
- Procesos infecciosos: Otite externa, otite das piscinas, ectima nasal etc.
- Procesos alérxicos, para o caso das gotas nasais. A rinite alérxica é unha patoloxía de alta prevalencia que en moitas ocasións non é controlable con tratamento sistémico.
- Procesos inflamatorios nasais (polipos nasais) ou óticos.
- Patoloxías dermatolóxicas que afectan a pel do oído, como o psoríase.
Respecto das contraindicacións haberá que ter en conta as seguintes consideracións:
- Gotas nasais: Evitar substancias que alteren a actividade dos cilios das células da mucosa nasal, como as substancias boricadas.
- Gotas óticas: No posible evitar as solucións oleosas e ter presente a posibilidade de que a membrana timpánica non estea indemne.
Calquera dos diferentes produtos sanitarios empregados para cubrir e protexer unha ferida.[23] A súa finalidade última é a de promover a cicatrización da ferida. En función a iso, as características que ha de reunir un bo apósito son:[24]
- Impermeabilidade aos xermes, partículas e auga.
- Capacidade de absorción.
- Favorecemento do pH ácido.
- Esterilidade.
- Permeabilidade aos gases.
- Proporcionar illamento térmico.[25]
A estas propiedades habería que engadirlles outras derivadas da interacción co organismo:[26]
- Elasticidade e flexibilidade.
- Baixa adherencia á ferida.
- Alto grao de cohesión.
- Non tóxico.
- Non alerxizante.
Percutánea ou transdérmica
[editar | editar a fonte]Malia o comentado respecto da dificultade para a absorción dos principios activos, sempre existe a posibilidade de que se absorba unha parte dos mesmos a través da pel. Este fenómeno dáse con máis intensidade nas zonas onde a pel está menos queratinizada (cara), onde se favorece a humidade (engurras corporais) ou en peles máis sensibles como a dos nenos. Por outra banda, en ocasións, si que nos interesa que o principio activo sexa absorbido para facer o seu efecto a nivel sistémico. Esta vía, coñécese como vía percutánea ou vía transdérmica e presenta unhas características especiais.
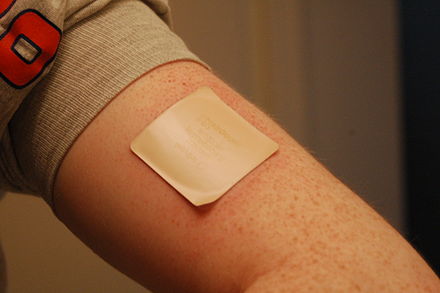
Os sistemas terapéuticos transdérmicos (STT) son formas de dosificación ideados para conseguir a contribución percutánea de principios activos a unha velocidade programada, ou durante un período de tempo establecido. Existen varios tipos de sistemas transdérmicos, entre os que se atopan:
Forma galénica consistente nun reservorio con principio activo que se libera lentamente ao aplicalo sobre a pel. Perséguese que o fármaco pase á circulación sistémica a través da pel e non a actividade do fármaco na propia pel. Estes parches proporcionan niveis plasmáticos terapéuticos constantes do fármaco, sempre que a pel permaneza intacta.[27] Aínda que relativamente recentes,[28] teñen numerosas indicacións e son obxecto de continua investigación.[27][29][30][31]
Técnica utilizada en fisioterapia que consiste na introdución dun fármaco ou principio activo (mediante a utilización de corrente galvánica ou outras formas de correntes derivadas desta) a través da pel.
Consiste na colocación sobre a pel de dous eléctrodos que, pola súa polaridade, fan que un fármaco cargado iónicamente atravese a pel. Prodúcese pola interacción da polaridade do eléctrodo coa carga iónica da substancia elixida (mediante o rexeitamento dos ións no polo do mesmo signo).
As vías de introdución a través da pel son sobre todo: folículos pilosos, glándulas sudoríparas e sebáceas, por interpor menor resistencia para o paso de substancias.
Deste xeito son administrados por vía percutánea fármacos cargados electricamente, sen producir efectos xerais no organismo, xa que os seus efectos son locais.
Inhalada
[editar | editar a fonte]
- Aerosois: son dispositivos que conteñen solucións ou suspensións dun principio activo, envasadas nun sistema a presión de maneira que, ao accionar a válvula, prodúcese a liberación do principio activo en forma de microgotas impulsado grazas a un axente propelente. Os principios activos que poden ser vehiculizados nesta forma galénica son numerosos:
- Agonistas β2 adrenérxicos:
- De Acción curta: salbutamol, levalbuterol, terbutalina e bitolterol.
- De acción prolongada: salmeterol, formoterol, bambuterol.
- Anticolinérxicos, como o bromuro de ipratropio.
- Glucocorticoides: beclometasona ou fluticasona.
O seu uso por vía inhalada oriéntanos cara ás patoloxías que se beneficiarían do tratamento cos aerosois:[32]

- Inhaladores de po seco: a partir do medicamento en estado sólido, libéranse partículas suficientemente pequenas de forma sincrónica coa inspiración; a forza da inhalación arrastra o produto.
- Nebulizadores: son dispositivos que fan pasar unha corrente de aire sobre un líquido que leva o principio activo en disolución. Xéranse partículas uniformes e moi finas do principio activo (líquido) que son inspiradas xunto co aire que inxecta o nebulizador. Este sistema permite que o fármaco penetre máis profundamente nas vías aéreas.
- Inhalacións.
- Insuflacións.
Innovacións galénicas
[editar | editar a fonte]A industria farmacéutica está continuamente desenvolvendo novas formas de facer chegar o fármaco ao seu destino da forma máis rápida e eficaz. Do mesmo xeito que o descubrimento de novos fármacos, as novas galénicas están suxeitas a patente de propiedade, pasando ao cabo dun tempo a dominio público. Así, por exemplo, Norman Leo Henderson e Louis Nasir Elowe inventaron en 1969 as cápsulas de liberación prolongada coa patente 3427378.[33] Deste xeito, continuamente estanse inventando sistemas, que baseados nas fórmulas galénicas clásicas, aportan un plus de efectividade ao produto comercial. Algúns exemplos serían:
- Sistema OROS ou de Microbomba osmótica. É un sistemas que permite a liberación temporal controlada do fármaco. Está constituído por un reservorio que contén o fármaco, formado por un núcleo sólido con capacidade osmótica. Rodeando o reservorio existe unha membrana semipermeable que permite o paso da auga procedente do exterior do sistema. Cando o comprimido entra en contacto co mollo gastrointestinal, a penetración da auga produce a disolución do núcleo osmótico e a saída do medicamento por un orificio ou zona de liberación. O tamaño do poro da membrana semipermeable vai condicionar a maior ou menor entrada de auga e, xa que logo, a velocidade de liberación do principio activo.[34]
- Sistema Filmtab.
- Sistema MUPS: En 1998 AstraZéneca desenvolveu o sistema MUPS (Multiple Unit Pellet System) ou Sistema Multigranular. A formulación dos comprimidos MUPS permite unha liberación rápida de 1.000 a 2.000 unidades de principio activo con protección fronte ao ácido no estómago. Son máis pequenas que as unidades contidas nunha cápsula tradicional, dispersanse con facilidade e disólvense no intestino delgado ofrecendo unha eficacia máis predecible.[35]
- Liposomas: Os liposomas son vesículas extraordinariamente pequenas compostas principalmente por fosfolípidos organizados en bicapas. Estas vesículas conteñen unha fase acuosa interna e están suspendidas nunha fase acuosa externa. Utilízanse basicamente para transportar os principios activos dun xeito o máis selectiva posible. Dependendo da súa natureza, o fármaco pódese incorporar dentro do liposoma (se é hidrofílico) ou na bicapa liposomal (caso dos lipófilos.[36] As vantaxes desta forma galénica serían:
- Aumento da eficacia e diminución da toxicidade do principio activo encapsulado.
- Prolongación do efecto.
- Mellor absorción, penetración e difusión.
- Posibilidade de vías de administración alternativas.
- Estabilización do principio activo.
- Sistema Flas: "É unha forma sólida oral que ten como propiedade que non fai falta deglutir o contido, senón que se absorbe directamente en contacto coa cavidade bucal"[11]
- Sistema Chronosphère.
- Xel Termoreversíbel : forma farmacéutica que permite que a preparación mantéñase en forma de solución líquida a temperaturas inferiores a 25 °C; en cambio a temperaturas próximas a 35 °C (cando entra en contacto co corpo humano) aumenta o seu viscosidade e a solución líquida transfórmase en gel. É ideal para uso en mucosas, xa que a forma líquida favorece a penetración pero ao converterse en sólido evítase o goteo e a deglutición. Os laboratorios españois SALVAT utilizan este sistema para algúns dos seus produtos de uso por vía nasal.[37]
Produción
[editar | editar a fonte]Os fármacos son producidos xeralmente pola industria farmacéutica. Os novos fármacos poden ser patentados, cando a empresa farmacéutica foi a que investigou e lanzado ao mercado o novo fármaco. Os dereitos de produción ou licenza de cada novo fármaco está limitado a un lapso que oscila entre 10 e 20 anos. Os fármacos que non están patentados chámanse fármacos copia; en cambio, aqueles que non están patentados pero teñen un estudo de bioequivalencia, aprobado polas autoridades locais, chámanse medicamentos xenéricos.
Estudos de estabilidade
[editar | editar a fonte]O obxectivo destes estudos é prover evidencia documentada de como as características físicas, químicas, microbiolóxicas e biolóxica do medicamento varían co tempo baixo a influencia de factores como temperatura, humidade, luz e establecer as condicións de almacenamento adecuadas e o período de caducidade.
A estabilidade dun fármaco pódese definir como a propiedade deste envasasado nun determinado material para manter durante o tempo de almacenamento e uso das características físicas, químicas, fisicoquímicas, microbiolóxicas e biolóxicas entre os límites especificados.
As formas de inestabilidade dun fármaco son:
- Química
Solvólosis, oxidación, deshidratación, racemización, incompatibilidades de grupo.
- Física
Polimorfismo, vaporización, adsorción.
- Biolóxica
Fermentación e xeración de toxinas
Datas de expiración
[editar | editar a fonte]- Artigo principal: Data de caducidade.
No ano 1979, nos Estados Unidos estableceuse unha lei que pedía ás empresas farmacéuticas que puxesen a data ata a cal garantíase a máxima potencia do medicamento. Noutras palabras, non indica o momento en que xa non é seguro tomalas nin cando se volven daninas. A maioría de medicinas son moi durables e degradanse lentamente, polo que a maioría, mentres sexa apropiadamente fabricada, manteñen a súa potencia en altos rangos moito despois da súa data de caducidade. Un executivo de Bayer, Chris Allen, dixo que as aspirinas que fabrican levan unha data de caducidade de tres anos posterior á súa fabricación, pero esta podería ser de ata máis e esta aínda estaría na súa máxima potencia.[Cómpre referencia] O motivo detrás disto xace en que constantemente están mellorando a fórmula das aspirinas, e facer probas de caducidade de máis de 4 anos por cada actualización sería irrazonable.
Con todo, rexistráronse problemas con medicamentos que inclúen tetraciclina; tamén hai excepcións na duración, como coa nitroglicerina, a insulina, e algúns antibióticos líquidos.[38][39]
Obxectivo terapéutico
[editar | editar a fonte]Os fármacos adminístranse co fin de conseguir un obxectivo terapéutico onde a concentración axeitada e a dose requirida para alcanzar este obxectivo dependen, entre outros factores, do estado clínico do paciente, a gravidade da patoloxía a tratar, a presenza doutros fármacos e de enfermidades intercorrentes.
Para iso requírese non soamente lograr unha resposta farmacolóxica e poder mantela; polo tanto, é necesario alcanzar a concentración apropiada do fármaco no lugar de acción e para iso é necesario coñecer a súa farmacocinética.
Debido ás diferenzas individuais, o tratamento eficaz require planificar a administración segundo as necesidades do paciente. Tradicionalmente, isto efectuábase por medio do axuste empírico da dose ata conseguir o obxectivo terapéutico.
Non obstante, a miúdo este método é pouco axeitado debido á demora en conseguir o efecto ou á aparición de toxicidade. Unha aproximación alternativa consiste en iniciar a administración de acordo coa absorción, distribución e eliminación, esperadas no paciente, e, posteriormente, axustar a dose segundo a resposta clínica e por medio da monitorización das concentracións plasmáticas.
Este enfoque require coñecer a farmacocinética do fármaco en función da idade, o peso e as consecuencias cinéticas das posibles enfermidades intercorrentes (renais, hepáticas, cardiovasculares ou unha combinación delas).
Parámetros farmacocinéticos básicos
[editar | editar a fonte]O comportamento farmacocinético da maioría dos fármacos pode resumirse por medio dalgúns parámetros. Os parámetros son constantes, aínda que os seus valores poden diferir dun paciente a outro e, no mesmo doente, en situacións distintas.
A biodispoñibilidade expresa o grao de absorción pola circulación sistémica.
A constante de absorción define a velocidade de absorción.
As modificacións destes dous parámetros inflúen sobre a concentración máxima ou pico, o tempo que tarda en alcanzarse a concentración máxima e a área baixo a curva (ABC) concentración-tempo tras unha dose oral única.
Nos tratamentos crónicos, o grao de absorción é a medida máis importante que se relaciona coa concentración media, mentres que o grao de flutuación depende da constante de absorción.
O volume aparente de distribución é o líquido teórico corporal en que tería que haberse disolto o fármaco para alcanzar a mesma concentración que no plasma. Úsase para saber a dose requirida para alcanzar unha concentración determinada no sangue. A fracción libre é útil porque relaciona a concentración total coa libre, que é quen, presumiblemente, está máis asociada cos efectos farmacolóxicos. É un parámetro útil sobre todo se se alterase a fixación ás proteínas plasmáticas, por exemplo en caso de hipoalbuminemia, de enfermidade renal e/ou hepática e en interaccións por desprazamento da unión a devanditas proteínas. O volume aparente de distribución e a fracción libre son os parámetros máis utilizados para o estudo da distribución do fármaco.
A velocidade de eliminación dun fármaco do organismo é proporcional á súa concentración plasmática. O parámetro que relaciona a ambas medidas é o aclaramento total ou clearance, que é a suma do aclaramento renal máis o aclaramento extrarrenal ou metabólico.
A fracción de fármaco eliminado sen cambios (en forma inalterada) é un parámetro útil para avaliar o efecto potencial das enfermidades renais e hepáticas sobre a eliminación dos fármacos. Unha fracción baixa indica que o metabolismo hepático é o mecanismo de eliminación e que unha enfermidade hepática podería afectar a eliminación do fármaco. As patoloxías renais producen maiores efectos na cinética de fármacos con elevada fracción de fármaco eliminado inalteradamente.
A velocidade con que se extrae un fármaco do sangue por un órgano excretor como o fígado non pode exceder a velocidade á que chega a devandito órgano. É dicir, o aclaramento presenta un valor límite. Cando a extracción é elevada, a eliminación está limitada pola chegada de fármaco ao tecido e, xa que logo, pola perfusión deste. Cando o órgano de eliminación é o fígado ou a parede intestinal e o fármaco adminístrase vía oral, unha porción da dose administrada pode ser metabolizada durante o seu paso obrigado a través dos tecidos ata a circulación sistémica; é o que se denomina efecto ou metabolismo de primeiro paso hepático.
Xa que logo, sempre que unha substancia teña unha extracción (aclaramento) elevada en fígado ou parede intestinal, a biodispoñibilidade por vía oral, será baixa, ata o punto que ás veces pode desaconsellar a administración vía oral, ou requirir a administración dunha dose oral moi superior á dose parenteral equivalente.
Algúns exemplos de fármacos presentan un efecto de primeiro paso importante;
- alprenolol,
- hidralazina,
- isoproterenol,
- lidocaína,
- meperidina,
- morfina,
- nifedipina,
- nitroglicerina,
- propranolol,
- testosterona,
- verapamilo.
A constante de eliminación está en función de como se elimina o fármaco do sangue por parte dos órganos excretores e como se distribúe polo organismo.
A vida media de eliminación beta, (ou semivida) é o tempo requirido para que a concentración plasmática -ou a cantidade de fármaco do organismo- se reduza nun 50%.
Para a maioría dos fármacos, a vida media mantense constante malia a cantidade de fármaco que estea presente no organismo, aínda que existen excepcións (difenilhidantoína, teofilina e heparina).
Tempo medio de permanencia (TMP) é outra medida da eliminación do fármaco. Defínese como o tempo medio que a molécula dun fármaco permanece no organismo tras unha administración intravenosa rápida. Do mesmo xeito que o aclaramento, o seu valor é independente da dose administrada. Para fármacos que describen un modelo de distribución monocompartimental, o TMP iguálase ao recíproco da constante de eliminación.
Variabilidade dos parámetros farmacocinéticos
[editar | editar a fonte]Identificáronse moitas das variables que afectan os parámetros farmacocinéticos, as cales deben terse en conta para axustar a dose administrada ás necesidades de cada paciente. Dado que ata despois do axuste das doses adoita persistir unha notable variabilidade, é necesaria unha monitorización coidadosa da resposta farmacolóxica e, en moitos casos, da concentración plasmática.
Idade e peso
[editar | editar a fonte]Para algúns fármacos establecéronse con precisión os cambios farmacocinéticos relacionados coa idade e o peso. Nos nenos e mozos (de 6 meses a 20 anos), a función renal correlacionase ben coa superficie corporal. Así, no caso dos fármacos que se eliminan principalmente en forma inalterada pola ouriña, o aclaramento varía coa idade de acordo co cambio da superficie corporal. Nas persoas maiores de 20 anos, a función renal diminúe aproximadamente un 1% ao ano. Tendo en conta estes cambios, é posible axustar a dose de devanditos fármacos en cada idade. Tamén se demostrou que a superficie corporal correlacionase co aclaramento metabólico nos nenos, aínda que hai moitas excepcións.
Nos recentemente nados e os lactantes, tanto a función renal como a hepática non están completamente desenvolvidas e non é posible facer xeneralizacións, agás que existen cambios rápidos.
Alteración da función renal
[editar | editar a fonte]O aclaramento renal da maioría dos fármacos varía proporcionalmente á depuración de creatinina, calquera que sexa a patoloxía renal. O cambio no aclaramento total depende da contribución dos riles na eliminación total de devandito fármaco. Xa que logo, espérase que o aclaramento total sexa proporcional á función renal (depuración de creatinina) para fármacos que se excretan exclusivamente de xeito inalterado e non resulte afectado no caso de fármacos que se eliminan por metabolismo.
Ás veces, na insuficiencia renal modifícase o volume aparente de distribución. No caso da digoxina, a redución do volume de distribución depende da menor fijación aos tecidos. No caso da difenilhidantoína, o ácido salicílico e moitos outros fármacos, o volume de distribución aumenta debido a que se reduce a fixación ás proteínas plasmáticas.
Nas situacións de estrés fisiolóxico (p.ex.,infarto de miocardio, cirurxía, colite ulcerosa ou enfermidade de Crohn) aumenta a concentración do reactante de fase aguda (1-glucoproteína ácida). En consecuencia, aumenta a fixación ás proteínas por parte dalgúns fármacos de natureza básica como o propranolol, a quinidina e a disopiramida; por conseguinte, diminúe o volume aparente de distribución destes fármacos.
As enfermidades hepáticas producen modificacións no aclaramento metabólico, pero non se dispón de bos predictores destes cambios. Describiuse unha redución espectacular da metabolización de fármacos na cirrose hepática. Nesta enfermidade a miúdo obsérvase unha diminución da fixación ás proteínas plasmáticas debido á menor concentración de albumina en sangue. Habitualmente, a hepatite aguda cursa con elevación dos enzimas séricos, por iso non se asocia a unha alteración do metabolismo.
Outras enfermidades como a insuficiencia cardíaca, a pneumonía, o hipertiroidismo e moitas outras enfermidades tamén poden alterar a farmacocinética.
Interaccións farmacolóxicas
[editar | editar a fonte]As interaccións farmacolóxicas provocan cambios nos valores dos parámetros farmacocinéticos e, xa que logo, na resposta terapéutica. Sábese que as interaccións afectan todos os parámetros. A maioría delas son graduais e a súa magnitude depende da concentración dos dous fármacos en interacción. Por todo iso, son difíciles de predicir e o axuste das doses é complicado.
Dependencia da dose
[editar | editar a fonte]En determinados casos, os valores dos parámetros farmacocinéticos varían segundo a dose administrada, a concentración plasmática ou o tempo; por exemplo, a diminución da biodispoñibilidade da griseofulvina ao aumentar a dose, debido a súa menor solubilidade nas secrecións do tracto gastrointestinais alto. Outro exemplo é o aumento desproporcionado da concentración en equilibrio estacionario da difenilhidantoína ao aumentar o intervalo de dosificación, porque os enzimas metabolizadores de difenilhidantoína teñen unha capacidade limitada para eliminar o fármaco e a velocidade de administración habitual aproxímase á velocidade máxima de metabolización. Para rematar, a redución da concentración plasmática de carbamazepina cando se administra de xeito crónica porque este fármaco induce o seu propio metabolismo.
Outras causas de variabilidade cinética dependente da dose son: saturación da fixación ás proteínas plasmáticas e tisulares (fenilbutazona), secreción saturable no ril (doses altas de penicilina) e metabolismo de primeiro paso hepático saturable (propranolol).
Os fármacos son case sempre compostos estraños ao organismo. Como tales, non se están formando e eliminando continuamente do mesmo xeito que sucede coas substancias endóxenas. Xa que logo, os procesos de absorción, biodispoñibilidade, distribución e eliminación teñen unha importancia capital para determinar o inicio, a duración e a intensidade do efecto farmacolóxico.
Absorción
[editar | editar a fonte]Proceso de transporte do fármaco desde o lugar de administración ata a circulación sistémica atravesando polo menos unha membrana celular.
A absorción dos fármacos vén determinada polas súas propiedades físico-químicas, formulacións e vías de administración.
As formas nas que se presentan os medicamentos (p. ex., píluras, comprimidos, cápsulas ou solucións) consisten no fármaco e outros ingredientes. Os medicamentos formúlanse para poder administralos por diversas vías (oral, bucal, sublingual, rectal, parenteral, tópica e inhalatoria). Un requisito esencial para que calquera fármaco poida absorberse é que sexa capaz de disolverse. Os medicamentos sólidos (p. ex., os comprimidos) poden disgregarse e desintegrarse, pero a absorción só ocorre cando o principio activo se disolve.
Transporte polas membranas celulares
[editar | editar a fonte]Cando os fármacos penetran no organismo a través da maioría das vías de administración (excepto a vía intravenosa ou intrarterial), deben atravesar varias membranas celulares semipermeábeis antes de chegar á circulación xeral.
Estas membranas actúan como barreiras biolóxicas que, de modo selectivo, inhiben o paso das moléculas do fármaco.
As membranas celulares compóñense fundamentalmente dunha matriz lipídica bimolecular que contén colesterol e fosfolípidos. Os lípidos proporcionan estabilidade á membrana e determinan as súas características de permeabilidade. Na matriz lipídica atópanse embutidas macromoléculas proteicas globulares de volume e composición variables. Algunhas destas proteínas da membrana participan no proceso de transporte e tamén poden ter a función de receptores para a regulación celular.
Os fármacos atravesan as barreiras biolóxicas por
- Difusión pasiva,
- Difusión facilitada
- Transporte activo
- Pinocitose.
Difusión pasiva
[editar | editar a fonte]Neste proceso, o transporte a través da membrana celular depende do gradiente de concentración do soluto. A maioría das moléculas pasan a través da membrana por difusión simple desde unha zona con elevada concentración (p. ex., líquidos gastrointestinais ) ata unha zona de baixa concentración (p. ex., o sangue). Posto que as moléculas do fármaco son rapidamente transportadas a través da circulación sistémica e distribúense enseguida nun gran volume de líquidos corporais e tecidos, a súa concentración no plasma é baixa ao principio, en comparación coa concentración no lugar de administración; este amplo gradiente é a forza impulsora do proceso. A velocidade neta de difusión é directamente proporcional a este gradiente, pero depende tamén da liposolubilidade, grao de ionización e tamaño molecular do fármaco e da superficie de absorción.
Con todo, posto que a membrana celular é de natureza lipídica, os fármacos liposolubles difunden con maior rapidez que aqueles relativamente non liposolubles. Ademais, as moléculas pequenas tenden a penetrar nas membranas con maior rapidez que as de maior volume.
A maioría dos fármacos son ácidos ou bases orgánicas débiles que no medio acuoso están de forma ionizada e non ionizada. A importancia deste concepto está en que a fracción non ionizada adoita ser liposoluble e difunde facilmente a través das membranas celulares. A forma ionizada non pode penetrar nas membranas tan facilmente, debido á súa baixa liposolubilidade e elevada resistencia eléctrica. A resistencia eléctrica é resultante da carga da molécula e dos grupos con carga eléctrica da superficie da membrana. Xa que logo, a penetración dun fármaco pode atribuírse principalmente á fracción non ionizada.
A distribución dun fármaco ionizable a través dunha membrana no equilibrio vén determinada polo pKa da substancia que é a inversa da constante de disociación (cando o pH e o pK son iguais, as concentracións da forma ionizada e non ionizada do fármaco son iguais) e polo gradiente de pH, se existe.
No caso dun ácido débil, canto maior sexa o pH, menor será o cociente entre a fracción non ionizada e a fracción ionizada. No plasma (pH = 7,4), a proporción entre as formas non ionizada e ionizada dun ácido débil (p. ex., cun pKa de 4,4) é 1:1.000; no mollo gástrico (pH = 1,4), a proporción invístese, é dicir, 1.000:1.
Por exemplo:
Cando o ácido débil, como a aspirina, se administra vía oral, o gradiente de concentración para a fracción non ionizada entre o estómago e o plasma tende a aumentar, situación que favorece a difusión a través da mucosa gástrica. Ao alcanzar o equilibrio, as concentracións de fármaco non ionizado no estómago e no plasma son idénticas porque só a forma non ionizada pode atravesar as membranas; a concentración de fármaco ionizado en plasma sería entón aproximadamente 1.000 veces superior á concentración de fármaco ionizado na luz gástrica. No caso dunha base débil cun pKa de 4,4, a situación é a inversa.
Así, os fármacos que son acedos débiles (p. ex., a aspirina), teoricamente deberían absorberse con maior facilidade nun medio ácido (como a luz gástrica), que as bases débiles (p. ex., a quinidina). Con todo, independentemente do pH do fármaco, a maior parte da absorción ten lugar no intestino delgado pola súa extensión.
Difusión facilitada
[editar | editar a fonte]Para certas moléculas (p.ex., glicosa), a velocidade de penetración é maior á esperada pola súa baixa liposolubilidade. Postúlase que existe un transportador que se combina de xeito reversible coa molécula substrato na parte externa da membrana celular e que o complexo transportador-substrato difundese rapidamente a través da membrana, liberando o substrato na superficie interna da membrana. Este proceso de difusión mediado por un transportador caracterízase pola selectividade e a saturabilidade. O transportador só acepta substratos cunha configuración molecular relativamente específica e o proceso está limitado pola dispoñibilidade de transportadores. Trátase dun mecanismo que non require enerxía, posto que o substrato non se transporta en contra dun gradiente de concentración.
Transporte activo
[editar | editar a fonte]Ademais da selectividade e da capacidade de saturación, o transporte activo caracterízase porque require gasto de enerxía por parte da célula. Os substratos poden acumularse no interior da célula contra gradiente de concentración. Os procesos de transporte activo están limitados aos fármacos con similitude estrutural coas substancias endóxenas. Estes fármacos adoitan absorberse en lugares específicos do intestino delgado. Identificáronse procesos de transporte activo para diversos ións, vitaminas, azucres e aminoácidos.
Pinocitose
[editar | editar a fonte]Consiste no englobamento e a captación de partículas ou líquido por parte dunha célula. A membrana celular se invagina, encerra á partícula ou ao líquido e logo volve fusionarse formando unha vesícula que máis tarde se desprende e emigra cara ao interior da célula. Este mecanismo tamén require gasto de enerxía. Probablemente, a pinocitose desempeña un papel menor no transporte de fármacos, coa excepción dos fármacos que son proteínas.
Características da administración oral
[editar | editar a fonte]Posto que a vía oral é o modo de administración máis frecuente, a absorción adoita referirse ao transporte dos fármacos a través das membranas das células epiteliales do tracto gastrointestinal.
A absorción trala administración oral depende das diferenzas do pH luminal ao longo do tubo dixestivo, da superficie de absorción, da perfusión tisular, da presenza de fluxo biliar e mucoso e das membranas epiteliais.
Os ácidos absórbense máis rapidamente no intestino que no estómago, en aparente contradición coa hipótese de que a forma non ionizada dun fármaco atravesa con maior facilidade as membranas. Con todo, esta discrepancia débese á enorme superficie do intestino delgado e á maior permeabilidade das súas membranas.
A mucosa oral posúe un epitelio delgado e moi vascularizado que favorece a absorción, pero o contacto sucede polo xeral por un tempo demasiado curto, ata para fármacos en solución, para que a absorción sexa apreciable. En ocasións, pode reterse o fármaco durante máis tempo, para que a absorción sexa máis completa, situando o fármaco entre a enxiva e o carrillo (administración bucal) ou colocándoo baixo a lingua ( sublingual ).
O estómago posúe unha superficie epitelial relativamente extensa, pero debido á grosa capa mucosa e a que o fármaco está en contacto relativamente pouco tempo, a absorción é limitada.
Dado que a absorción de practicamente todos os fármacos é máis rápida no intestino delgado que no estómago, a velocidade de baleirado gástrico é o paso limitante. Os alimentos, especialmente os graxos, enlentecen o vaciamento gástrico (e a velocidade de absorción); este feito explica por que se recomenda tomar algúns fármacos co estómago baleiro cando se desexa que a acción comece rapidamente. A presenza de alimentos pode aumentar a absorción se o fármaco é pouco soluble (p. ex., a griseofulvina); reducila, se o fármaco se degrada no estómago (p. ex., a penicilina G), ou ter un efecto moi pouco significativo ou nulo. Ademais, os principios activos que alteran o vaciamento gástrico (p. ex., os parasimpaticolíticos) tamén afectan a velocidade de absorción doutros fármacos.
O intestino delgado posúe a maior superficie para a absorción no tracto gastrointestinal. No duodeno o pH intraluminal oscila entre 4 e 5, pero vólvese progresivamente máis alcalino ao longo do tubo dixestivo (na porción distal do íleo é próximo a 8). A flora gastrointestinal pode inactivar determinados fármacos, reducindo así a súa absorción e a súa biodispoñibilidade. A redución do fluxo sanguíneo (p. ex., no shock) pode diminuír o gradiente de concentración a través da mucosa intestinal e reducir a absorción por difusión pasiva. (A diminución do fluxo sanguíneo periférico tamén altera a distribución e o metabolismo dos fármacos.)
A velocidade do tránsito intestinal pode influír na absorción, especialmente no caso de fármacos que se absorben por medio de transporte activo (p. ex., as vitaminas do complexo B), fármacos que se disolven lentamente (p. ex., a griseofulvina) ou os que son demasiado polares (pouco liposolubles) para atravesar con facilidade as membranas (p. ex., moitos antibióticos). Para este tipo de fármacos, o tránsito debe ser moi lento para que a absorción sexa completa.
Para as formas medicamentosas de liberación controlada, a absorción pode ocorrer inicialmente no intestino groso, especialmente cando a liberación do principio activo da forma medicamentosa dura máis de 6 h, que é o tempo de tránsito do intestino groso.
Absorción de solucións
[editar | editar a fonte]A absorción dos fármacos que se administran vía oral en forma de solución depende de si estes son capaces de sobrevivir aos «encontros» perigosos coas numerosas secrecións gastrointestinais, os baixos pH e as encimas potencialmente degradadoras. Polo xeral, ata se un fármaco é estable no ambiente intestinal, unha escasa fracción del pasa ao intestino groso. Fármacos escasamente lipófilos (de baixa permeabilidade), como os aminoglicósidos, absórbense lentamente da solución no estómago e no intestino delgado; para estes fármacos, a absorción polo intestino groso suponse que será ata máis lenta, xa que a superficie de absorción é menor. En consecuencia, estes fármacos non son candidatos para formas de liberación controlada.
Absorción de formas sólidas
[editar | editar a fonte]A maioría dos fármacos que se administran vía oral preséntanse en forma de comprimidos ou cápsulas, sobre todo por economía, estabilidade e aceptación por parte do paciente. Antes de absorberse débense desintegrar e disolver. A desintegración aumenta considerablemente a superficie do fármaco que entra en contacto cos líquidos gastrointestinais e facilita a súa disolución e a súa absorción.
A miúdo, durante o proceso de fabricación do medicamento engádense os desintegrantes e outros excipientes (como disolventes, lubricantes, surfactantes, fixadores e dispersantes) para facilitar o proceso de desintegración.
Os surfactantes aumentan a velocidade de disolución do fármaco ao incrementar a súa humectación, solubilidade e dispersabilidade. Entre os factores que modifican ou atrasan a desintegración das formas sólidas figuran a excesiva presión exercida na elaboración do comprimido e a aplicación de recubrimentos especiais para protexelo dos procesos dixestivos gastrointestinais. Os lubricantes hidrófobos (p. ex., o estearato de Mg) poden fixar o principio activo e reducir a súa biodispoñibilidade.
A velocidade de disolución determina a cantidade de fármaco dispoñible para a absorción. Cando é máis lenta que o proceso de absorción, a disolución constitúe o paso limitante e pode manipularse por medio de cambios na formulación do produto.
A miúdo emprégase a redución do tamaño das partículas para aumentar a superficie do fármaco, o cal resulta eficaz para aumentar a velocidade e a magnitude da absorción gastrointestinal dun fármaco no que estes parámetros están limitados pola súa lenta disolución. Entre os factores que afectan á velocidade de disolución están, se o fármaco está en forma de sal, en forma cristalina ou en hidrato. Por exemplo, os sales sódicos dos ácidos débiles (como barbitúricos e salicilatos) disólvense máis rapidamente que os seus ácidos correspondentes, calquera que sexa o pH do medio. Certos fármacos son polimorfos, podendo existir en forma amorfa ou en varias formas cristalinas. O palmitato de cloranfenicol existe en dúas formas, pero só unha posúe un grao suficiente de disolución para que a súa absorción sexa de utilidade clínica. Cando unha ou máis moléculas de auga se combinan cun fármaco en forma cristalina constitúese un hidrato. A solubilidade do hidrato pode ser moi distinta da que posúe a forma non hidratada do composto; así, a ampicilina anhidra ten maior velocidade de disolución e de absorción que o seu trihidrato correspondente.
Administración parenteral
[editar | editar a fonte]A administración directa dun fármaco no torrente circulatorio (habitualmente por vía intravenosa asegura a chegada de toda a dose á circulación xeral. Con todo, a administración do fármaco por unha vía que requira o seu paso a través dunha ou máis membranas biolóxicas (inxección intramuscular ou s.c.) para alcanzar o sangue non garante que se absorba totalmente. Para fármacos proteicos con PM >20.000 g/mol, o paso a través das membranas dos capilares é tan lento que trala administración intramuscular ou subcutanea a maior parte da absorción realízase a través do sistema linfático «por defecto». Nestes casos, a velocidade de liberación á circulación sistémica é lenta e incompleta, xa que hai un fenómeno de metabolismo de primeiro paso polos enzimas proteolíticos dos vasos linfáticos.
Como os capilares tenden a ser moi porosos, a perfusión (fluxo sanguíneo por gramo de tecido) é o factor determinante da velocidade de absorción no caso de moléculas pequenas. Xa que logo, o lugar da inxección inflúe na absorción do fármaco; así, a velocidade de absorción do diazepam inxectado por vía intramuscular nunha zona con escaso fluxo sanguíneo pode ser moito máis lenta que trala administración dunha dose vía oral
Cando se inxectan sales de ácidos ou bases pouco solubles por vía intramuscular, é posible que a absorción se atrase ou sexa errática. Por exemplo, a forma parenteral de difenilhidantoína (fenitoína) é unha solución do sal sódico en propilenglicol ao 40%, cun pH próximo a 12. Cando se inxecta por vía intramuscular, o propilenglicol absórbese e os líquidos hísticos, actuando como tampón, reducen o pH e producen un desprazamento do equilibrio entre a forma ionizada e o ácido libre. É entón cando o ácido libre, que é pouco soluble, precipita. En consecuencia, a disolución e a absorción son moi lentas (entre 1 e 2 semanas).
Formas de liberación controlada
[editar | editar a fonte]As formas de liberación sostida deseñáronse co fin de reducir a frecuencia de administración e as flutuacións nas concentracións plasmáticas e, desta forma, conseguir un efecto farmacolóxico uniforme. Ademais, a menor frecuencia de administración é máis cómoda para o paciente e pode mellorar o cumprimento da prescripción. En principio, os fármacos apropiados para este tipo de formas farmacéuticas son os que requiren unha dosificación frecuente debido á súa curta vida media de eliminación e á breve duración do seu efecto.
A miúdo, as formas orais de liberación sostida están deseñadas para manter concentracións terapéuticas do fármaco durante 12 h ou máis. A velocidade de absorción pode controlarse por distintas vías: recubrindo as partículas do fármaco con ceras ou outras substancias insolubles en auga, incluíndo ao principio activo nunha matriz da que se libera lentamente ao longo do tracto gastrointestinal ou elaborando un complexo entre o fármaco e resinas de intercambio iónico.
As formas tópicas de liberación sostida deseñáronse para a presenza do fármaco durante períodos prolongados. Por exemplo, a difusión da clonidina a través dunha membrana proporciona unha liberación sostida do fármaco durante 1 semana, e un polímero impregnado de nitroglicerina fixado a un vendaxe adhesivo proporciona unha liberación controlada durante 24 h. Os fármacos de liberación transdérmica deben posuír características adecuadas para a penetración cutánea e unha potencia elevada, xa que a velocidade de penetración e a área de absorción son limitadas.
Formuláronse moitos preparados de administración parenteral non intravenosa, co fin de proporcionar concentracións plasmáticas sostidas. No caso de antibióticos, a inxección intramuscular de sales insolubles (p. ex., a penicilina G benzatina) consegue efectos farmacolóxicos clinicamente útiles durante longos períodos de tempo. Outros fármacos están formulados como suspensións ou solucións en vehículos non acuosos. Así, por exemplo, a insulina pode inxectarse como suspensión cristalina para conseguir un efecto prolongado; a insulina amorfa, cunha superficie de disolución maior, posúe un inicio do efecto rápido e unha duración curta.
Bioequivalencia e Biodispoñibilidade
[editar | editar a fonte]Biodispoñibilidade é a proporción de principio activo (fármaco ou metabolito) que entra na circulación xeral e que, por conseguinte, chega ao lugar de acción, así como a velocidade con que iso sucede.
Mentres que as propiedades fisicoquímicas dun fármaco condicionan o seu potencial de absorción, as propiedades da forma farmacéutica (do seu deseño e da súa manufactura) son determinantes principais da súa biodispoñibilidade. As diferenzas na biodispoñibilidade entre diferentes formulacións dun mesmo fármaco poden ser clinicamente relevantes.
O concepto de equivalencia entre as formulacións dun fármaco é importante á hora de decidir o tratamento máis adecuado en cada situación.
O termo equivalente químico (ou farmacéutico) refírese aos medicamentos que conteñen o mesmo principio activo na mesma cantidade e que cumpren os estándares oficiais; con todo, os ingredientes inactivos dos medicamentos poden ser distintos.
A palabra bioequivalencia aplícase aos equivalentes químicos que, administrados á mesma acode seguindo a mesma pauta, alcanzan concentracións similares no plasma e nos tecidos. Os equivalentes terapéuticos designan dous medicamentos que, administrados á mesma persoa e coa mesma pauta, proporcionan esencialmente o mesmo efecto terapéutico ou tóxico. Suponse que os medicamentos bioequivalentes son terapeuticamente equivalentes.
Con frecuencia, algúns dos problemas terapéuticos (p. ex., toxicidade, perda de eficacia) que ocorren no curso de tratamentos prolongados, cando a enfermidade estaba sendo controlada cunha formulación dun fármaco, son debidos ao cambio por sustitutos non equivalentes (como no caso da digoxina ou a difenilhidantoína).
En ocasións é posible a equivalencia terapéutica aínda que haxa variacións na biodispoñibilidade. Por exemplo, o índice terapéutico (relación entre a dose máxima tolerada e a dose mínima eficaz) da penicilina é tan amplo que diferenzas moderadas na concentración plasmática debidas a diferenzas na biodispoñibilidade das formulacións de penicilina poden non afectar a eficacia terapéutica ou a seguridade do fármaco. Pola contra, se se tratase dun fármaco cun índice terapéutico relativamente estreito, as diferenzas na biodispoñibilidade si serían determinantes.
A biodispoñibilidade tamén depende doutros factores, como os relacionados coa fisioloxía e as patoloxías -principal e asociadas- do paciente.
Este concepto é fundamental para explicarse por que as drogas non teñen sempre a mesma magnitude de efecto
A velocidade á que se absorbe un fármaco é un factor importante incluso cando o fármaco se absorbe totalmente. Pode ocorrer que sexa demasiado lenta para alcanzar unha concentración plasmática terapéutica ou tan rápida que se alcancen concentracións tóxicas tras cada dose.
Causas de baixa biodispoñibilidade
[editar | editar a fonte]Cando un fármaco se disolve rapidamente da súa formulación e atravesa as membranas con facilidade, a absorción tende a ser completa na maioría das vías de administración. Este non sempre é o caso dos fármacos administrados vía oral Antes de alcanzar a vea cava, un fármaco debe descender polo tracto gastrointestinal e atravesar a parede intestinal e o fígado, que son lugares onde habitualmente se metabolizan os fármacos; xa que logo, é posible que o fármaco se metabolice antes de chegar á circulación xeral. Esta causa de baixa biodispoñibilidade denomínase metabolismo de primeiro paso. Moitos fármacos teñen unha baixa biodispoñibilidade debido a que sofren un elevado metabolismo de primeiro paso. En moitos casos (p. ex., isoproterenol, noradrenalina, testosterona), a extracción neses tecidos é tan completa que a biodispoñibilidade é practicamente cero. Con todo, para fármacos con metabolitos activos, as consecuencias terapéuticas deste efecto de primeiro paso dependen da contribución do fármaco ou do metabolito aos efectos farmacolóxicos desexables ou tóxicos.
A biodispoñibilidade escasa é frecuente en formulacións para administración oral de fármacos pouco solubles en auga, que se absorben moi lentamente. Cando a absorción é lenta ou incompleta, os factores que poden afectar a biodispoñibilidade son máis numerosos que cando é rápida ou completa. No primeiro caso, pode esperarse unha resposta terapéutica moito máis variable.
A permanencia insuficiente do fármaco no tracto gastrointestinal é unha das causas máis comúns de biodispoñibilidade escasa. Ao inxerir un fármaco, este non permanece no tracto gastrointestinal máis de 1-2 días e no intestino delgado só se acha 2-4 horas. Se o fármaco non se disolve con facilidade ou se é incapaz de atravesar o epitelio intestinal (fármacos polares, moi ionizados), o tempo no que permanece no lugar de absorción pode ser insuficiente. Nestes casos, a biodispoñibilidade non só é baixa, senón que tende a ser moi variable. Ademais, outros factores como idade, sexo, actividade, fenotipo xenético, estrés, enfermidades (p. ex., aclorhidria, síndromes de mala absorción) ou a cirurxía gastrointestinal previa, poden alterar e ata aumentar aínda máis as diferenzas na biodispoñibilidade.
As reaccións que compiten coa absorción poden reducir a biodispoñibilidade. Poden ser a formación de complexos (p. ex., entre tetraciclina e ións metálicos polivalentes), a hidrólise debida ao ácido gástrico ou aos enzimas dixestivos (p. ex., a hidrólise da penicilina e o palmitato de cloranfenicol), a conxugación na parede intestinal (p. ex., a sulfo-conxugación do isoproterenol), a adsorción por outros fármacos (p. ex., digoxina e colestiramina) e o metabolismo pola microflora intestinal.
Estimación da biodispoñibilidade
[editar | editar a fonte]A análise da biodispoñibilidade a partir de datos sobre a concentración plasmática respecto ao tempo adoita requirir tres medidas: a concentración plasmática máxima (pico) do fármaco, o tempo en que aparece esta concentración máxima e a área baixo a curva concentración plasmática-tempo
A concentración plasmática aumenta ao facelo a velocidade e o grao de absorción; cando a velocidade de eliminación do fármaco equivale á velocidade de absorción, alcánzase o pico.
As determinacións da biodispoñibilidade baseadas na concentración plasmática máxima poden ser erróneas, posto que a eliminación empeza inmediatamente despois de que o fármaco chega á circulación. O tempo en que se alcanza o pico plasmático depende da velocidade de absorción; de feito, é o índice máis utilizado para medir este parámetro. Canto máis lenta sexa a absorción, máis tarde se alcanzara o pico.
Con todo, a miúdo o pico máximo non é un índice absolutamente definitivo, xa que se trata dun valor puntual, que depende da frecuencia na toma nas mostras de sangue e, no caso de que as concentracións próximas ao pico describan unha curva relativamente plana, da reproducibilidade dos resultados. Recordar que a concentración sanguínea dunha droga moitas veces non é índice adecuado da concentración no sitio de acción.
Dose única vs. doses múltiples
[editar | editar a fonte]Aínda que para medir a velocidade de absorción a administración única proporciona máis datos que as administracións múltiples, a biodispoñibilidade pode medirse tras administracións únicas ou tras administracións repetidas.
O estudo dos parámetros farmacocinéticos tras dose múltiple posúe algunhas vantaxes, como permitir representar de xeito máis exacto a situación clínica habitual. Normalmente conséguense concentracións plasmáticas superiores ás obtidas con dose única, o que facilita a súa determinación.
Tras administrar doses repetidas con intervalos regulares durante o período correspondente a 4-5 vidas media beta ou de eliminación, as concentracións plasmáticas deberían alcanzar o estado de equilibrio estacionario (a cantidade de fármaco absorbido iguálase coa cantidade de fármaco eliminado en cada intervalo de dose).
No caso dos fármacos que se excretan en forma inalterada (sen metabolizar) pola ouriña, pode estimarse a biodispoñibilidade medindo a cantidade total de fármaco excretado tras unha administración única. Idealmente, debería recollerse a ouriña durante un período correspondente a 7-10 vidas medias beta de eliminación co fin de recuperar por completo o fármaco absorbido. En caso de múltiples doses, a biodispoñibilidade pode determinarse medindo o fármaco inalterado en ouriña durante 24h en condicións de equilibrio estacionario.
Distribución
[editar | editar a fonte]Tras chegar á circulación xeral, o fármaco pasa aos tecidos do organismo. Polo común a distribución é desigual polas diferenzas na perfusión sanguínea, o grao de unión aos tecidos, as variacións rexionais do pH e a distinta permeabilidade das membranas celulares.
A velocidade de penetración do fármaco no tecido depende do fluxo sanguíneo, da masa de tecido e da proporción do fármaco en sangue e no tecido.
Nas zonas cunha vascularización rica alcánzase o equilibrio de distribución (a velocidade de entrada e a velocidade de saída son iguais) entre o plasma e o tecido máis rapidamente que nas zonas pouco perfundidas, a non ser que a difusión a través das membranas sexa un paso limitante. Tras alcanzar o equilibrio de distribución, as concentracións do fármaco (libre e unida a proteínas, véxase máis adiante) nos tecidos e no líquido extracelular quedan reflectidas pola concentración plasmática. O metabolismo e a excreción teñen lugar simultaneamente coa distribución, o que determina un proceso dinámico e complexo.
Volume aparente de distribución
[editar | editar a fonte]O volume de líquido no que parece distribuírse ou diluírse o fármaco denomínase volume aparente de distribución (o volume corporal en que tería que disolverse o fármaco para alcanzar a mesma concentración que no plasma). Este parámetro informa sobre a concentración plasmática esperada para unha dose concreta e tamén sobre a dose requirida do fármaco para obter unha concentración concreta. Con todo, o volume aparente de distribución non proporciona datos sobre o patrón específico de distribución. Cada fármaco distribúese no organismo dun modo particular. Algúns tenden a dirixirse aos tecidos graxos, outros permanecen no líquido extracelular e, para rematar, outros fixanse con avidez a tecidos específicos, como o fígado ou o ril.
Moitos fármacos acedos (p. ex., a warfarina e o ácido salicílico) fíxanse moito a proteínas e, xa que logo, teñen un volume aparente de distribución pequeno. Moitos fármacos básicos (como a anfetamina e a meperidina) son captados con avidez polos tecidos e o seu volume de distribución é maior que o volume de todo o organismo.
Fármaco unido ligado a proteínas
[editar | editar a fonte]O grao de distribución dos fármacos nos tecidos depende da súa unión ás proteínas plasmáticas e a diversos compoñentes tisulares.
Unión a proteínas plasmáticas
[editar | editar a fonte]Os fármacos son transportados no sangue en parte en solución (como fármaco libre, non unido) e en parte fixados a diversos compoñentes do sangue (proteínas e células sanguíneas).
O principal determinante da proporción entre o fármaco unido e o fármaco libre é a interacción reversible entre o fármaco e a proteína á que se fixa; esta interacción segue a lei de acción de masas.
Moitas proteínas plasmáticas poden interaccionar cos fármacos. As máis importantes son a albumina, a glucoproteína ácida a1 e as lipoproteínas. Os fármacos de natureza ácida adoitan fixarse á albumina, en tanto que os de tipo básico tenden a unirse a unha das dúas últimas ou a ambas
Posto que só o fármaco libre pode sufrir unha difusión pasiva cara aos tecidos e as zonas extravasculares nas que se exerce o efecto farmacolóxico, a concentración de fármaco libre reflicte mellor a concentración do fármaco no lugar de acción e, xa que logo, os seus efectos.
A fracción libre (proporción de fármaco libre en relación á concentración total) é un parámetro máis útil que a fracción unida. A fixación ás proteínas plasmáticas inflúe na distribución e na relación aparente entre a actividade farmacolóxica e a concentración plasmática total do fármaco. A concentracións elevadas de fármaco, a cantidade de fármaco unido aproxímase a un límite máximo, dependendo do número de sitios de unión dispoñibles.
Xa que logo, dise que a fixación é saturable. A saturabilidade é a base das interaccións por desprazamento entre fármacos.
Fixación aos tecidos
[editar | editar a fonte]Os fármacos poden unirse a moitas substancias, ademais das proteínas. Esta unión pode ser moi específica, como é o caso da fixación da cloroquina aos ácidos nucleicos. A unión tisular adoita involucrar a asociación do fármaco cunha macromolécula nun medio acuoso. Outro tipo de asociación que induce a pensar nunha fixación tisular é a distribución do fármaco na graxa corporal. Dado que o tecido adiposo está pouco perfundido, o tempo necesario para alcanzar o equilibrio nel é prolongado.
Reservorio de fármacos
[editar | editar a fonte]A acumulación nos tecidos ou nos compartimentos corporais pode prolongar a permanencia dos fármacos no plasma e as súas accións porque os tecidos serven de depósito. A medida que a concentración plasmática diminúe, o fármaco almacenado vaise liberando á circulación. A localización do lugar de acción e as diferenzas relativas na distribución tisular tamén poden ser importantes. O indutor anestésico tiopental, un tiobarbitúrico, é un exemplo de fármaco cuxo almacenamento en reservorios tisulares inicialmente acurta o seu efecto farmacolóxico, pero tras administracións repetidas prolóngao. O tiopental é un hipnótico moi liposoluble e distribúese rapidamente no cerebro trala inxección intravenosa única. A concentración no cerebro aumenta en 1 á 2 minutos, e posteriormente diminúe rapidamente, no cerebro e máis lenta a concentración plasmática. A hipnose finaliza a medida que o fármaco se redistribue desde o cerebro ao plasma e cara aos tecidos con perfusión máis lenta, músculo e graxa. Con todo, se se determinan as concentracións plasmáticas durante o tempo suficiente, pode observarse unha terceira fase de distribución que representa a liberación lenta do fármaco acumulado no tecido adiposo. A administración continua de tiopental supón que grandes cantidades de fármaco almacénanse no tecido graxo, o cal prolonga o efecto hipnótico.
Algúns fármacos acumúlanse nas células en concentracións superiores ás alcanzadas no líquido extracelular. Esta acumulación adoita implicar a fixación de fármacos a proteínas celulares, fosfolípidos ou ácidos nucleicos. Os antipalúdicos como a cloroquina destacan pola súa notable fixación intracelular, de modo que poden alcanzar concentracións intracelulares en leucocitos e células hepáticas miles de veces superiores ás plasmáticas. O fármaco almacenado atópase en equilibrio co plasmático e volve ao plasma a medida que se vai eliminando do organismo.
Barreira hematoencefálica
[editar | editar a fonte]Os fármacos chegan ao sistema nervioso central (SNC) pola circulación capilar e a través do líquido cefalorraquídeo (LCR). Aínda que o cerebro recibe unha proporción importante do volume minuto (aproximadamente 1/6), a distribución dos fármacos no cerebro está restrinxida. Algúns fármacos liposolubles (como o tiopental) entran e exercen os seus efectos rapidamente, pero moitos outros -en particular os máis hidrosolubles- penetran no cerebro con maior lentitude. As células endoteliales dos capilares cerebrais están máis estreitamente unidas entre si que as dos demais leitos capilares do organismo; isto contribúe á lenta penetración das substancias hidrosolubles. Outra barreira importante para os fármacos hidrosolubles son as células do tecido glial (os astrocitos) que forman unha vaina pegada á membrana basal do endotelio capilar.
O endotelio capilar e a vaina astrocítica constitúen a barreira hematoencefálica. Esta barreira é a que confire as características diferenciais de permeabilidade entre estes tecidos e os do resto do organismo, nos que a barreira corresponde á parede capilar e non á célula parenquimatosa. Así, os compostos polares son incapaces de penetrar no cerebro, pero poden acceder ao líquido intersticial da maioría dos demais tecidos. O concepto de barreira hematoencefálica definiuse trala observación de que os colorantes polares podían penetrar na maioría dos tecidos, pero non no SNC.
Os fármacos poden pasar directamente ao LCR ventricular a través do plexo coroide, e teñen acceso ao tecido cerebral por difusión pasiva desde o LCR. O plexo coroide tamén é unha zona de transporte activo de ácidos orgánicos (como a penicilina) desde o LCR ao sangue.
Os factores principais que determinan a velocidade de penetración no LCR ou noutras células son o grao de fixación ás proteínas, o grao de ionización e o cociente de partición lípido/auga do composto. A velocidade de penetración no cerebro é lenta nos fármacos que se unen en gran proporción a proteínas. No caso de ácidos e bases débiles ionizados, a penetración é tan lenta que se considera practicamente inexistente.
Noutros tecidos do organismo, a perfusión é o determinante principal da velocidade de distribución, pero o SNC está tan ben perfundido que o factor máis importante adoita ser a permeabilidade. Con todo, nos tecidos pouco perfundidos (p. ex., o músculo e o tecido adiposo), a distribución prolóngase notablemente, sobre todo se o tecido ten moita afinidade polo fármaco.
Eliminación e/ou segregación
[editar | editar a fonte]Suma de procesos que conducen á desaparición (por metabolismo e excreción) do fármaco do organismo.
Metabolismo
[editar | editar a fonte]O fígado é o órgano principal onde se produce o metabolismo dos fármacos (modificacións químicas), pero non é o único. Algúns metabolitos teñen actividade farmacolóxica. Cando a substancia administrada é inactiva pero dá lugar a un metabolito activo, o composto administrado denomínase profármaco, especialmente se foi deseñado para liberar eficazmente o principio activo.
Reaccións metabólicas
[editar | editar a fonte]O metabolismo dos fármacos supón un amplo espectro de reaccións químicas:
- oxidación,
- redución,
- hidrólise,
- hidratación,
- conxugación,
- condensación
- isomerización.
Os enzimas implicados nestas reaccións están presentes en numerosos tecidos, pero, polo xeral, atópanse máis concentradas no fígado. Para moitos fármacos, o metabolismo prodúcese en dúas fases. As reaccións de fase I supoñen a formación dun novo grupo funcional ou unha partición da molécula (oxidación, redución, hidrólise); trátase de reaccións non sintéticas.
As reaccións de fase II comportan a conxugación cun composto endóxeno (p. ex., ácido glucurónico, sulfato, glicina); trátase, pois, de reaccións sintéticas. Os metabolitos formados nas reaccións sintéticas son máis polares e máis facilmente excretados polo ril (en ouriños) e polo fígado (na bile) que os formados nas reaccións non sintéticas. Algúns fármacos sofren procesos de metabolismo de ambos tipos. Pese a que se denominan fases I e II, trátase, como pode verse, dunha clasificación funcional, non secuencial, das reaccións de metabolismo de fármacos.
Citocromo P-450
[editar | editar a fonte]O sistema enzimático máis importante do metabolismo de fase I é o citocromo P-450, unha superfamilia de enzimas microsomais que catalizan reaccións de oxidación de numerosos fármacos pola súa capacidade de transferencia de electróns.
Os electróns son achegados pola NADPH-citocromo P-450-redutase, unha flavoproteína que transfire electróns do NADPH (a forma reducida do fosfato dinu-cleótido de nicotinamida-adenina) ao citocromo P-450.[40] Os enzimas do citocromo P-450 están agrupados en 14 familias de xenes de mamífero que comparten secuencias idénticas e 17 subfamilias. Denomínanse por un símbolo raíz (CYP), seguido dun numeral árabe para a familia, unha letra para a subfamilia e outro número árabe para o xene específico.
Os enzimas das subfamilias 1A, 2B, 2C, 2D e 3A son os máis importantes do metabolismo en mamíferos. CYP1A2, CYP2C9, CYP2C19, CYP2D6 e CYP3A4 son os máis importantes no metabolismo humano. A especificidade dos enzimas permite explicar moitas interaccións entre fármacos. Na táboa 298-3 preséntanse varios exemplos de fármacos que interaccionan con enzimas específicos do complexo citocromo P-450 (v. tamén Interaccións farmacolóxicas, cap. 301). As diferenzas xenéticas entre pacientes poden modificar a resposta clínica.
Conxugación
[editar | editar a fonte]A glucuronoconxugación é a reacción de fase II máis común, e é a única que ocorre no sistema enzimático microsomal hepático. Os glicurónidos se segregan pola biles e elimínanse polos ouriños. O cloranfenicol, o meprobamato e a morfina son algúns exemplos de fármacos metabolizados por esta vía.
A conxugación con aminoácidos, como a glutamina e a glicina, produce metabolitos (p. ex., ácido salicilúrico, da conxugación de ácido salicílico e glicina) facilmente excretables nos ouriños, pero que non adoitan secretarse pola bile. A acetilación é a vía metabólica principal das sulfamidas. A hidralazina, a isoniazida e a procainamida tamén sofren acetilación. A sulfoconxugación é a reacción entre grupos fenol ou alcohol e un sulfato inorgánico, que deriva en parte de aminoácidos que conteñen sofre como a cisteína. Os ésteres de sulfato así obtidos son polares e se excretan rapidamente nos ouriños. Algúns exemplos de fármacos que forman sulfatos son: paracetamol, estradiol, metildopa, minoxidil e tiroxina. A metilación é a principal vía metabólica para inactivar algunhas catecolaminas. A niacinamida e o tiouracilo tamén sofren procesos de metilación.
Modificacións debidas á idade
[editar | editar a fonte]Os recentemente nados teñen un sistema enzimático microsomal hepático só parcialmente desenvolvido e, en consecuencia, presentan algunhas dificultades para metabolizar moitos fármacos (p. ex., hexobarbital, fenazetina, anfetamina e clorpromazina). A experiencia co cloranfenicol en recentemente nacidos mostra claramente as graves consecuencias que se poden derivar do enlentecemento da glucuronoconxugación. Doses equivalentes en mg/kg de cloranfenicol, ben toleradas por pacientes maiores, poden provocar unha toxicidade grave nos recentemente nados (síndrome do neno gris), asociada á presenza de niveis plasmáticos elevados de cloranfenicol durante longo tempo.
A miúdo, a capacidade metabólica tamén se atopa diminuída nos pacientes anciáns; esta redución varía en función do fármaco e non é tan grave como nos recentemente nados.
Conservación e cuidados dos medicamentos
[editar | editar a fonte]
A conservación adecuada dos medicamentos é fundamental para manter a súa actividade farmacolóxica de forma óptima. En xeral, deben evitarse os lugares accesibles aos nenos, para evitar intoxicacións, e os lugares moi húmidos, moi cálidos ou á intemperie, para evitar a degradación do principio activo (fármaco). Convén observar a data de caducidade que aparece na embalaxe, así como as instrucións de conservación indicadas no prospecto.[41]
Reciclado
[editar | editar a fonte]Unha vez terminado o tratamento para o que foron prescritos, os medicamentos deben depositarse nun punto limpo, posto que desvotalos indiscriminadamente xunto co resto dos residuos pode deteriorar gravemente o medio ambiente.
Iniciativa SIGRE (España)
[editar | editar a fonte]Co obxectivo de pechar correctamente o ciclo de vida dos medicamentos, a industria farmacéutica puxo en marcha SIGRE Medicamento e Medio Ambiente (Sistema Integrado de Xestión e Recollida de Envases), en colaboración coa farmacias e a distribución do sector, facilitando que os cidadáns poidan desprenderse comodamente, pero con todas a garantías sanitarias e medioambientais, dos restos de medicamentos e dos seus envases a través do Punto SIGRE situado nas oficinas de farmacia.
Normativa
[editar | editar a fonte]España
[editar | editar a fonte]A información que aparece na etiquetaxe dos medicamentos, tanto da embalaxe exterior como do acondicionamento primario, así como os símbolos, siglas e lendas que deben conter, defínese nos anexos III e IV do Real Decreto 1345/2007, do 11 de outubro, polo que se regula o procedemento de autorización, rexistro e condicións de dispensación dos medicamentos de uso humano fabricados industrialmente.[42][43][44][45] [46] [47]
Notas
[editar | editar a fonte]- ↑ Definicións no Dicionario da Real Academia Galega e no Portal das Palabras para fármaco.
- ↑ Definicións no Dicionario da Real Academia Galega e no Portal das Palabras para medicamento.
- ↑ "US Federal Food, Drug, and Cosmetic Act, SEC. 210., (g)(1)(B)." (en inglés). Arquivado dende o orixinal o 12-05-2009. Consultado o 16-12-2012.
- ↑ "Directive 2004/27/EC of the European Parliament and of the Council of 31 March 2004 amending Directive 2001/83/EC on the Community code relating to medicinal products for human use. Article 1." (en inglés). Consultado o 16-12-2012.
- ↑ "agent medicinal". L'Enciclopèdia.cat. Barcelona: Grup Enciclopèdia Catalana.
- ↑ Albarracín,A. et. al. 1.984 Historia del medicamento. Vol. I. Ed. Doyma S.A. Barcelona. 99 pp.
- ↑ Definicións no Dicionario da Real Academia Galega e no Portal das Palabras para electuario.
- ↑ Medicina na beira interior da prehistoria ao século XXI (PDF) (en portugués). p. 19. Arquivado dende o orixinal (PDF) o 06 de marzo de 2016. Consultado o 16 de marzo de 2014.
- ↑ Mª del Carmen Francés Causapé La Colección de medicamentos. Arquivado 11 de agosto de 2011 en Wayback Machine.(en castelán)
- ↑ Lastres, J.L. Director do Departamento de Farmacia e Tecnoloxía Farmacéutica. Facultade de Farmacia - Universidade Complutense de Madrid. na páxina web da Facultade de Farmacia e Bioquímica da Universidade de Buenos Aires. Arquivado 18 de abril de 2010 en Wayback Machine.
- ↑ 11,0 11,1 11,2 Domínguez-Gil,Alfonso. Catedrático de Tecnoloxía Farmacéutica da Universidade de Salamanca, en declaracións a revista Estar bien Arquivado 16 de marzo de 2014 en Wayback Machine. edición dixital, 6 de xuño de 2008, nº 60
- ↑ Lei 29/2006 de Garantías e Uso Racional dos Medicamentos e Produtos Sanitarios, en España.
- ↑ Medicamento. Diccionario de la Lengua Española (22ª ed.). Madrid: Real Academia Española; 2001. (en castelán)
- ↑ 14,0 14,1 Glosario de Presentaciones Farmacéuticas 2006. Dispoñible en Cofepris. Consultado o 22/06/2013
- ↑ Hernández Herrero, Gonzalo et al. MediPharm. Tratado de Medicina Farmacéutica. Editorial Médica Panamericana, 2010. ISBN 8498350107, 9788498350104
- ↑ "Pharmaceuticals". Arquivado dende o orixinal o 17 de xuño de 2013. Consultado o 21 de xullo de 2013.
- ↑ 17,0 17,1 17,2 17,3 Dulanto, F. Dermatología médico-quirúrgica. Tomo II. Cap. 53. 1.341 pp. Ed. Anel s.a. Granada. 1.982 ISBN 84-85622-18-9
- ↑ Información Técnica y Comercial del Agua alibour (en castelán)
- ↑ Sarkany, I T. St. John's Hospital Dermatol. Soc., 59,241,1.973
- ↑ Korting, G.W. Manual de Dermatología Ed. Científico-Médica Barcelona 1986 28 pp ISBN 84-224-0813-9
- ↑ Leppard, B. Ashton, R. Tratamiento en Dermatología. Radcliffe Medical Press. Oxford. 1994. 5 pp ISBN 1-85775-003-9
- ↑ 22,0 22,1 Galante, G.R. Solucións para mucosas. Dispoñible en [1] Arquivado 09 de xuño de 2007 en Wayback Machine. Consultado o 12 de outubro de 2008
- ↑ Enrich, L. y Negre, S. Propuesta de clasificación de los apósitos estériles modernos. Cienc. Pharm. 1998 8(4): 153-171
- ↑ Hall, V.,Murillo, N.,Quesada,M. (marzo de 2001). Centro de Información de Medicamentos, ed. Apósitos hidrocoloides. Su papel en la curación de heridas. (PDF) (en castelán). Arquivado dende o orixinal (PDF) o 09 de marzo de 2016. Consultado o 3 de xaneiro do 2016.
- ↑ INSALUD. Guía práctica clínica: selección y utilización de medicamentos en las residencias geriátricas. 2000. España Dispoñible en [2] Arquivado 28 de xullo de 2011 en Wayback Machine.
- ↑ Serna, J. Vitales, M. López, M.C., Molina, A. Tomo II Capítulo 4: Dermatología en Farmacia Hospitalaria 868 pp, dispoñible en [3] Arquivado 05 de decembro de 2008 en Wayback Machine.
- ↑ 27,0 27,1 Marcotegui Ros, F. Sistemas terapéuticos transdérmicos. Boletín de información farmacoterapéutica de Navarra. Vol 1 nº 3. 1993. Dispoñible en [4]
- ↑ Food and Drug Administration (ed.). "FDA approves scopolamine patch to prevent peri-operative nausea" (en inglés). Consultado o 12 de febreiro de 2007.
- ↑ Rodriguez, R., Daza, P. y Rodriguez, M. F. Uso de buprenorfina transdérmica en el alivio del dolor por cáncer. Rev. Col. Anest. [online]. Oct./Dec. 2006, vol.34, no.4 [revisada o 10 de outubro de 2008], p.253-257. Dispoñible en [5]. ISSN 01203347.
- ↑ Clasificación Internacional de Patentes CIP 2007. Véxase en
- ↑ "Memoria L. Novartis 2008. Dispoñible en" (PDF). Arquivado dende o orixinal (PDF) o 20 de marzo de 2009. Consultado o 01 de novembro de 2013.
- ↑ British Thoracic Society & Scottish Intercollegiate Guidelines Network (SIGN). British Guideline on the Management of Asthma. Guideline No. 63. Edinburgh:SIGN; 2004. (HTML Arquivado 18 de xuño de 2006 en Wayback Machine., Full PDF Arquivado 24 de xullo de 2006 en Wayback Machine., Summary PDF Arquivado 24 de xullo de 2006 en Wayback Machine.)
- ↑ "Patente europea".
- ↑ Formas farmacéuticas y vias de administración de fármacos. Arquivado 03 de decembro de 2008 en Wayback Machine. Departamento de Farmacología y Terapéutica. Facultad de Medicina. Universidade Autónoma de Madrid.
- ↑ El último avance en terapia gastrointestinal. El primer IBP desarrollado como isómero en Diariosalud.net Arquivado 23 de setembro de 2015 en Wayback Machine.
- ↑ "Véxase Conceptos básicos sobre Liposomas en". Arquivado dende o orixinal o 26 de setembro de 2008. Consultado o 24 de maio de 2014.
- ↑ "Nasolina". Salvat (en castelán). Consultado o 2022-04-10.
- ↑ Richard Altschuler "DO MEDICATIONS REALLY EXPIRE?" Redflagsdaily, 2002. Republicación en Medscape
- ↑ Jasińska M et al "Stability studies of expired tablets of metoprolol tartrate and propranolol hydrochloride" Journal Acta Pol Pharm, 2009. PMID 20050534
- ↑ Kenneth Jensen e Birger Lindberg Møller (febreiro 2010). "Plant NADPH-cytochrome P450 oxidoreductases". Phytochemistry (en inglés) 71 (2–3): 132–41. PMID 19931102. doi:10.1016/j.phytochem.2009.10.017. Consultado o 12 de xullo do 2020.
O CPR demostrouse que estaba localizado no retículo endoplasmático a principios dos anos 60 (Williams e Kamin, 1962).
- ↑ Casamitjana Nuria. Centro de Información del Medicamento. Colegio de Farmacéuticos de Barcelona, ed. "Medicamentos. Cómo conservarlos" (en castelán). Arquivado dende o orixinal o 24 de marzo de 2011. Consultado o 16 de decembro de 2016.
- ↑ "Larrañaga Arregui B. Medicamentos de uso humano: etiquetado y prospecto, novedades del Real Decreto 1345/2007. Sendagaiak. 2008; 21(2):7-8." (PDF). Arquivado dende o orixinal (PDF) o 17 de agosto de 2011. Consultado o 16 de decembro de 2016.
- ↑ Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano fabricados industrialmente. BOE. 2007/11/07; nº 267.
- ↑ Real Decreto 1718/2010, de 17 de diciembre, sobre receta médica y órdenes de dispensación. BOE. 2011/01/20; (17):6306-29.
- ↑ Real Decreto-ley 9/2011, de 19 de agosto, de medidas para la mejora de la calidad y cohesión del sistema nacional de salud, de contribución a la consolidación fiscal, y de elevación del importe máximo de los avales del Estado para 2011. BOE. 2011/08/20; (200):93143-68.
- ↑ Información que deberán incluir los prospectos de los medicamentos autorizados por la Agencia Española de Medicamentos y Productos Sanitarios
- ↑ Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) (23 de febreiro do 2012). "Instrucciones para la comunicación de aspectos del etiquetado susceptibles de provocar errores de medicación. Nueva dirección de correo electrónico" (PDF) (en castelán). Consultado o 6 de outubro do 2017.
Véxase tamén
[editar | editar a fonte]| Wikimedia Commons ten máis contidos multimedia na categoría: Fármaco |
Bibliografía
[editar | editar a fonte]- Finkelstein, Stan; Temin, Peter (2008). Reasonable Rx: Solving the Drug Price Crisis (en inglés). FT Press. ISBN 0-13-234449-1.
Ligazóns externas
[editar | editar a fonte]- Agencia Española de Medicamentos y Productos Sanitarios
- Fichas técnicas de los medicamentos en España
- Diagnosia.com - Información de medicamentos
- Lista modelo de medicamentos esenciais da OMS-2008
- Listaxe de Medicamentos Autorizados en España (uso humano)
- Organización Farmacéutica Colegial Arquivado 26 de abril de 2012 en Wayback Machine.
- SIGRE Medicamento y Medio Ambiente
- Vademecum de medicamentos Arquivado 21 de xullo de 2013 en Wayback Machine.
- Vademecum.com - Recursos sobre medicamentos
- Vademecum.es - Información de medicamentos