Enfermedad de Fabry
| Enfermedad de Fabry | ||
|---|---|---|
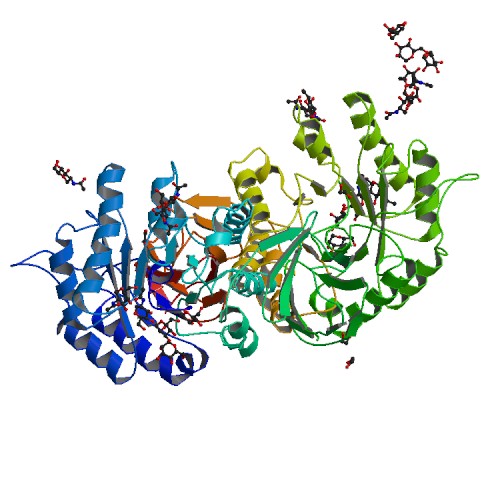 Alfa-galactosidasa, la proteína deficitaria en la enfermedad de Fabry. | ||
| Especialidad |
endocrinología nefrología dermatología cardiología | |
| Diagnóstico diferencial | Miocardiopatía hipertrófica | |
| Sinónimos | ||
| ||
La enfermedad de Fabry es una enfermedad de almacenamiento lisosómico hereditaria ligada al cromosoma X derivada de mutaciones en el gen que codifica la enzima α-galactosidasa. Fue descrita por Johannes Fabry y William Anderson en 1898. Se incluye dentro de las lipidosis o enfermedades por almacenamiento de lípidos.[1]
Epidemiología
[editar]Afecta más a los hombres que a las mujeres: se calcula que 1 en 40 000-60 000 varones recién nacidos tienen la enfermedad de Fabry. Sin embargo, la prevalencia en mujeres es desconocida. Esto último, sumando al hecho de que hay muchos casos de presentación tardía, ha ocasionado nuevas estimaciones sobre la enfermedad, que concluyen que la prevalencia en la población general es más cercana a 1 en 3000 personas.[2]
Causas
[editar]
La enfermedad de Fabry es un trastorno hereditario, cuya alteración genética (mutación) se localiza en el cromosoma X. El ADN, el bloque estructural de las células, está compuesto por 23 pares de cromosomas. La pareja cromosómica que determina el sexo contiene dos cromosomas X en el caso de las mujeres o un cromosoma X y otro Y en el de los hombres. Las hijas (XX) heredan un cromosoma X de la madre y un cromosoma X del padre. Por eso, todas las hijas de un hombre afectado heredarán un cromosoma X anómalo del padre y un cromosoma X normal de la madre. La presencia de un cromosoma X normal y otro alterado hace que los síntomas de la enfermedad de Fabry no siempre aparezcan o que, si lo hacen, suelan tardar más que en los varones. Los síntomas de la enfermedad de Fabry también pueden ser menos graves que en los varones, por lo que, a veces, es más difícil reconocer la enfermedad en las mujeres. Todos los hijos de un hombre afectado heredarán al cromosoma Y de su padre y el cromosoma X de su madre y, en consecuencia, no sufrirán la enfermedad.
En una situación, en la que la madre padezca la enfermedad de Fabry, la probabilidad de que la descendencia también la padezca es del 50 %. Si los hijos varones heredan el cromosoma X alterado de la madre, manifestarán los síntomas, pero si heredan el cromosoma X normal de la madre, no lo harán. Si las hijas heredan el cromosoma X anómalo, podrán, o no, desarrollar los síntomas de la enfermedad de Fabry.
Fisiopatología
[editar]La deficiencia lisosomal de alfa-galactosidasa A (alfa-Gal A) es el principal defecto metabólico.[1]
La herencia es recesiva ligada al cromosoma X, por lo que suele ser más grave en varones, ya que las mujeres pueden presentar uno de los cromosomas X normal y los síntomas serían más leves.
La expresión del fallo genético se traduce en el déficit de la enzima α-galactosidasa A, que se codifica normalmente en el brazo largo del cromososma X (Xq21.3–q23/24). El gen afectado es el de la α-galactosidasa (GLA), del cual se conocen 272 mutaciones.
Este defecto se manifiesta en la dificultad para metabolizar una substancia grasa, la globotriosilceramida o trihexosidogalactosilglucosil ceramido (abreviado como Gb3 o GL-3) en los lisosomas, produciéndose la acumulación generalizada de este metabolito, razón de la afección sistémica de distintos órganos.
La secreción y recaptación de la alfa galactosidasa provee las bases para la terapia de sustitución enzimática exógena de enzima en la terapéutica.
Los signos y síntomas se deben a la acumulación de G3b en las paredes de pequeños vasos, túbulos renales, células glomerulares, nervios y ganglios dorsales
En el caso de las mujeres transmisoras, lo asintomático es lo frecuente, la manifestación fenotípica de enfermedad más frecuente en ellas es la opacidad corneal.
Historia natural de la enfermedad
[editar]Como la progresión de la enfermedad de Fabry varía de una persona a otra, los síntomas pueden aparecer también a edades diferentes y con una intensidad diversa. De aquí la importancia que tiene evaluar los diferentes síntomas y entender su repercusión en las distintas etapas de la vida.
- Lactantes: De los múltiples síntomas de la enfermedad de Fabry, los primeros que suelen aparecer entre los niños pequeños son el dolor y las molestias relacionadas con el calor.
- Niños y adolescente: Además de los episodios de dolor la sensación de ardor en las manos y pies, los niños y los jóvenes con enfermedad de Fabry suelen mostrar una erupción de color rojo oscuro, moteada (angioqueratomas), que se acentúa más entre el ombligo y las rodillas; además, se observan cambios en el aspecto de la córnea que puede reconocer un óptico durante una exploración sistemática del ojo con un dispositivo especial. Los padres y los profesores deben plantearse los efectos del esfuerzo físico, del ejercicio y de las temperaturas extremas para los niños con enfermedad de Fabry. Asimismo hay que tener en cuenta cuestiones de tipo social relacionadas con la escuela o el empleo y vale la pena consultar a los especialistas.
- Adultos: La enfermedad de Fabry progresa lentamente. Los síntomas causados por el daño de los riñones, el corazón y el sistema nervioso central suelen aparecer entre los 30 y los 45 años. A veces, sólo cuando se realizan las pruebas para averiguar la causa de los problemas cardíacos o renales se descubre la acumulación de Gb3 y se establece el diagnóstico de la enfermedad de Fabry.
Cuadro clínico
[editar]La afectación se refleja en el terreno neurológico, gastrointestinal, cardíaco, renal, dermatológico y oftalmológico. Existe un período de latencia importante (años o décadas) entre el inicio de los síntomas y el del diagnóstico.
Clínicamente hay lesiones cutáneas muy típicas (angioqueratoma), disminución o ausencia de sudoración, opacidades en córnea y cristalino (cornea verticillata), dolor y parestesias en las extremidades, con afección vascular de distintos órganos como cerebro, corazón y riñón.
Se inicia en la infancia y adolescencia, con dolor y angioqueratomas. Los angioqueratomas son lesiones puntiformes de color rojo oscuro o azuladas, planas y simétricas, que no desaparecen con la presión. Aparecen con mayor frecuencia en la región entre el ombligo y las rodillas, pero pueden aparecer en cualquier parte de la piel y en las mucosas.
Los episodios de dolor quemante o urente en manos y pies (acroparestesias), pueden ser desencadenados por cambios en la temperatura, fiebre, ejercicio o fatiga y durar entre horas y días. Puede presentarse dolor abdominal (retortijones abdominales), que se confunde con apendicitis o cólicos renales.
Entre la segunda y cuarta décadas de la vida se presentan problemas renales y cardíacos. Los primeros manifestados como proteinuria, isostenuria y fallo renal progresivo, con hipertensión arterial sistémica. Los problemas cardíacos suelen ser la hipertrofia del ventrículo izquierdo, dolor precordial, infarto agudo del miocardio e insuficiencia cardíaca congestiva.
Edema de miembros inferiores sin hipoproteinemia, diarrea, náusea, tinnitus, y anhidrosis (ausencia de sudoración) son manifestaciones habituales, las que aumentan en número y gravedad con la edad.
Los síntomas más frecuentes son: acroparestesias, angioqueratomas, anhidrosis y córnea verticilada.
La causa de fallecimiento suele relacionarse a insuficiencia renal, insuficiencia cardiaca o enfermedad cerebrovascular.
Diagnóstico
[editar]En los hombres, el método más eficiente y confiable para diagnosticar la enfermedad de Fabry es un examen de sangre para la demostración de la deficiencia de la actividad de enzima alfa-galactosidasa, o alfa-GAL, en el plasma, leucocitos, lágrimas o fibroblastos de la piel.
Tratamiento
[editar]- Tratamiento de la enfermedad
Actualmente la enfermedad de Fabry no tiene cura, pero se ha conseguido un tratamiento de sustitución enzimática con agalsidase alpha (replagal) y agalsidase beta (fabrazyme), que consiste en reponer la enzima a-Gal A deficitaria y prevenir, así, la acumulación continua de Gb3.
La terapia con enzima no es curativa, requiere la aplicación endovenosa de la enzima cada dos semanas. El principal problema con este tratamiento es el costo, que llega hasta los 170 000 USD (unos 130 000 €) al año por cada paciente.
- Tratamiento de los síntomas
Hay que combatir los diferentes signos y síntomas de la enfermedad de Fabry para evitar las molestias y preservar la calidad de vida. El tratamiento sintomático varía en cada caso; suele utilizarse carbamazepina y fenitoína para reducir el dolor. La insuficiencia renal se trata con diálisis o trasplante renal. Además hay numerosos cuidados que puede hacer tanto el paciente como su cuidador para aliviar los síntomas:
- Dieta: Las molestias digestivas se pueden reducir tomando comidas pequeñas y más frecuentes.
- Entorno: El dolor relacionado con la enfermedad de Fabry puede desencadenarse por factores tales como la exposición al sol, los cambios bruscos de temperatura, el ejercicio físico o el estrés.
- Trabajo: Evitar los trabajos que exijan mucha habilidad manual, cambios rápidos de temperatura, ejercicio físico o estrés.
- Escuela: Los niños con enfermedad de Fabry pueden sentirse diferentes de sus compañeros y no siempre disfrutan plenamente de las actividades físicas. Por eso, conviene que los profesores y los compañeros sepan que su hijo padece la enfermedad de Fabry.
- Bienestar: Las enfermedades pueden producir dolor y empeorar los síntomas, de manera que las visitas regulares al médico para vigilar la salud general representan una parte importante del tratamiento global.
Véase también
[editar]Otras lipidosis:
- Enfermedad de Gaucher
- Enfermedad de Niemann-Pick
- Enfermedad de Wolman
- Xantomatosis cerebrotendinosa
- Sitosterolemia
- Enfermedad de Refsum
- Enfermedad de Tay-Sachs
- Leucodistrofia metacromática[3]
Referencias
[editar]- ↑ a b Bokhari, Syed Rizwan A.; Zulfiqar, Hassam; Hariz, Anis (2022). Fabry Disease. StatPearls Publishing. Consultado el 26 de julio de 2022.
- ↑ Germain DP. Enfermedad de Fabry [Internet]. 2012. 2012 [cited 2018 Oct 3]. Available from: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=324&lng=ES
- ↑ Manual Merck de información médica para el hogar. Capitulo 139: Alteraciones del colesterol y otras grasas. Consultado el 1-7-2010
Bibliografía
[editar]- Enfermedad de Fabry asociada a artritis reumatoide. Una encrucijada multisistémica. An Med Interna (Madrid). 2003; 20(1).
- ARIAS MARTÍNEZ FJ, BARBADO HERNÁNDEZ G, PÉREZ MARTÍN C, PÉREZ DE AYALA V, CASAL ESTEBAN JJ, VÁZQUEZ RODRÍGUEZ. Evaluación de pacientes con enfermedad de Fabry en la Argentina. Medicina (B. Aires). 2010; 70(1).
Enlaces externos
[editar]
| Control de autoridades |
|
|---|
Datos: Q615645
Multimedia: Fabry disease / Q615645