Paramyotonia congenita
| Paramyotonia congenita | |
|---|---|
| Other names | Paramyotonia congenita of von Eulenburg or Eulenburg disease[1] |
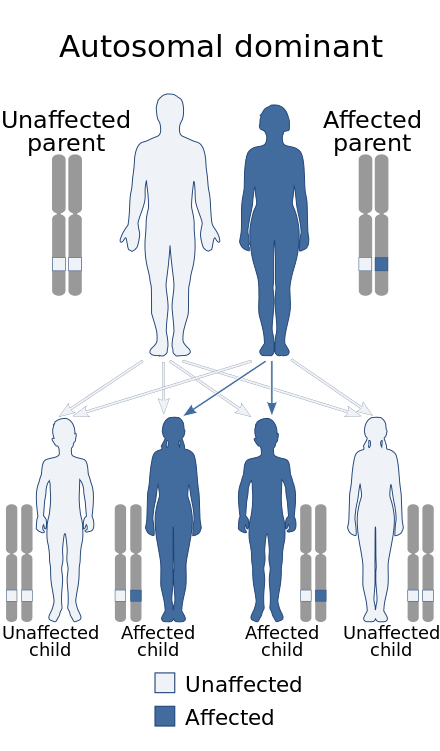 | |
| This condition is inherited in an autosomal dominant manner. | |
| Specialty | Neurology |
Paramyotonia congenita (PC) is a rare congenital autosomal dominant neuromuscular disorder characterized by "paradoxical" myotonia.[2] This type of myotonia has been termed paradoxical because it becomes worse with exercise whereas classical myotonia, as seen in myotonia congenita, is alleviated by exercise. PC is also distinguished as it can be induced by cold temperatures. Although more typical of the periodic paralytic disorders, patients with PC may also have potassium-provoked paralysis. PC typically presents within the first decade of life and has 100% penetrance. Patients with this disorder commonly present with myotonia in the face or upper extremities. The lower extremities are generally less affected. While some other related disorders result in muscle atrophy, this is not normally the case with PC. This disease can also present as hyperkalemic periodic paralysis and there is debate as to whether the two disorders are actually distinct.[3]
Symptoms and signs
[edit]Patients typically complain of muscle stiffness that can continue to focal weakness. This muscle stiffness cannot be walked off, in contrast to myotonia congenita. These symptoms are increased (and sometimes induced) in cold environments. For example, some patients have reported that eating ice cream leads to a stiffening of the throat. For other patients, exercise consistently induces symptoms of myotonia or weakness. Typical presentations of this are during squatting or repetitive fist clenching. Some patients also indicate that specific foods are able to induce symptoms of paramyotonia congenita. Isolated cases have reported that carrots and watermelon are able to induce these symptoms. The canonical definition of this disorder precludes permanent weakness in the definition of this disorder. In practice, however, this has not been strictly adhered to in the literature.[citation needed]
Pathophysiology
[edit]Paramyotonia congenita (as well as hyperkalemic periodic paralysis and the potassium-aggravated myotonias) is caused by mutations in a sodium channel, SCN4A. The phenotype of patients with these mutations is indicated in Table 1. These mutations affect fast inactivation of the encoded sodium channel. There are also indications that some mutations lead to altered activation and deactivation. The result of these alterations in channel kinetics is that there is prolonged inward (depolarizing) current following muscle excitation. There is also the introduction of a "window current" due to changes in the voltage sensitivity of the channel’s kinetics. These lead to a general increase in cellular excitability,[citation needed] as shown in figure 1.

There has been one study of a large number of patients with paramyotonia congenita. Of 26 kindreds, it found that 17 (71%) had a mutation in SCN4A while 6 (29%) had no known mutation. There is no large difference between these two groups except that patients with no known mutation have attacks precipitated less by cold but more by hunger, are much more likely to have normal muscle biopsies, and show less decreased compound muscle action potentials when compared to patients with known mutations.[5]
| Mutation | Region | Myotonia | Weakness | References | ||||
|---|---|---|---|---|---|---|---|---|
| Cold | Exercise/ Activity |
Potassium | Cold | Exercise/ Activity |
Potassium | |||
| R672C | D2S4 | ? | ? | ? | ? | ? | ? | [5] |
| I693T | D2S4-S5 | N | ? | ? | Y | Y | Y | [6] |
| T704M* | D2S5 | Y | ? | ? | Y | Y | Y | [7],[8],[9],[10] |
| S804F** | D2S6 | Y | Y | Y | ? | Y | N | [11] |
| A1152D | D3S4-S5 | Y | ? | ? | ? | ? | ? | [12] |
| A1156T* | D3S4-S5 | Y | ? | ? | ? | Y | ? | [3],[11] |
| V1293I | D3S4 | Y | Y | N | ? | ? | N | |
| G1306V** | D3-4 | Y | Y | Y | ? | ? | Y | [13],[14] |
| T1313A | D3-4 | Y | Y | N | Y | Y | N | [15] |
| T1313M | D3-4 | Y | Y | N | Y | Y**** | N | [13],[16] |
| M1360V* | D4S1 | ? | ? | ? | Y | Y | ? | [17] |
| M1370V* | D4S1 | Y | Y | N | N | N | Y | [18] |
| L1433R | D4S3 | Y | Y | Y | ? | Y***** | N | [16] |
| R1448C | D4S4 | Y | Y | N | N | Y | N | [6],[10],[19],[20] |
| R1448H | D4S4 | Y | Y | Y | Y | Y | ? | [10],[16],[19],[20] |
| R1448P | D4S4 | Y | Y | ? | Y | ? | N | [21] |
| R1448S | D4S4 | Y | Y | N | ? | Y | N | [22] |
| R1456E | D4S4 | Y | Y | N | N | N | N | [23] |
| V1458F*** | D4S4 | ? | ? | ? | ? | ? | ? | [24] |
| F1473S*** | D4S4-S5 | ? | ? | ? | ? | ? | ? | [24] |
| M1592V* | D4S6 | Y | Y | Y | Y | Y | Y | [10],[16],[25],[26],[27],[28],[29] |
| E1702K | C-term | ? | ? | N | ? | ? | N | [5] |
| F1795I | C-term | Y | ? | ? | ? | ? | ? | [30] |
* ** *** **** ***** |
Symptoms of both PC and hyperKPP (Periodica paralytica paramyotonica) Also diagnosed as a Potassium-aggravated myotonia Original case reports unpublished. When exercised in a cold environment After muscles were cooled | |||||||
| This table was adapted from Vicart et al., 2005.[31] "Cold" refers to symptoms either occurring or significantly worsening with cold temperatures. Likewise, "Exercise/Activity" refers to symptom onset or severity worsening with exercise and/or more general movement like hand clenching. "Potassium" refers to ingestion of food high in potassium or other disorders which are known to increase serum potassium levels. Mutation region nomenclature is: domain number (e.g., D1) followed by segment number (e.g., S4). Thus, D2S3 indicates that the mutation is in the 3rd membrane spanning loop of the 2nd domain. Some mutations occur between segments and are denoted similarly (e.g., D4S4-S5 occurs between the 4th and 5th segments of the 4th domain). Other mutations are located between domains and are denoted DX-Y where X and Y are domain numbers. C-term refers to the carboxy-terminus. | ||||||||
Diagnosis
[edit]Diagnosis of paramyotonia congenita is made upon evaluation of patient symptoms and case history. Myotonia must increase with exercise or movement and usually must worsen in cold temperatures. Patients that present with permanent weakness are normally not characterized as having PC. Electromyography may be used to distinguish between paramyotonia congenita and myotonia congenita.[32],[33] Clinicians may also attempt to provoke episodes or myotonia and weakness/paralysis in patients in order to determine whether the patient has PC, hyperkalemic periodic paralysis, or one of the potassium-aggravated myotonias. Genomic sequencing of the SCN4A gene is the definitive diagnostic determinant.[citation needed]
Treatment
[edit]Some patients do not require treatment to manage the symptoms of paramyotonia congenita. Others require treatment for their muscle stiffness and often find mexiletine to be helpful. Others have found acetazolamide to be helpful as well.[34] Avoidance of myotonia triggering events is also an effective method of myotonia prevention.[citation needed]
Epidemiology
[edit]Paramyotonia congenita is considered an extremely rare disorder, though little epidemiological work has been done. Prevalence is generally higher in European-derived populations and lower among Asians. Epidemiological estimates have been provided for the German population. There, it was estimated that the prevalence of PC is between 1:350,000 (0.00028%) and 1:180,000 (0.00056%).[20] However, the German population of patients with PC is not uniformly distributed across the country. Many individuals with PC herald from the Ravensberg area in North-West Germany, where a founder effect seems to be responsible for most cases.[20][35] The prevalence here is estimated at 1:6000 or 0.017%.[citation needed]
History
[edit]Originally thought to be separate from hyperkalemic periodic paralysis and the sodium channel myotonias, there is now considerable disagreement as to whether these disorders represent separate entities or overlapping phenotypes of a complex disorder spectrum. It was once thought that paramyotonia congenita was more common in males. Observation of the most recent generation has shown this to be untrue. On average, half of children in a family inherit the disorder regardless of gender.[36]
References
[edit]- ^ Facts About Myopathies | MDA Publications Archived 2007-08-05 at the Wayback Machine
- ^ Eulenburg A (1886). "Über eine familiäre durch 6 Generationen verfolgbare Form kongenitaler Paramyotonie". Neurol. Zentralbl. 12: 265–72.
- ^ a b de Silva S, Kuncl R, Griffin J, Cornblath D, Chavoustie S (1990). "Paramyotonia congenita or hyperkalemic periodic paralysis? Clinical and electrophysiological features of each entity in one family". Muscle Nerve. 13 (1): 21–6. doi:10.1002/mus.880130106. PMID 2325698. S2CID 24805255.
- ^ Cannon S, Brown R, Corey D (1993). "Theoretical reconstruction of myotonia and paralysis caused by incomplete inactivation of sodium channels". Biophys J. 65 (1): 270–88. Bibcode:1993BpJ....65..270C. doi:10.1016/S0006-3495(93)81045-2. PMC 1225722. PMID 8396455.
- ^ a b c Miller T, Dias da Silva M, Miller H, Kwiecinski H, Mendell J, Tawil R, McManis P, Griggs R, Angelini C, Servidei S, Petajan J, Dalakas M, Ranum L, Fu Y, Ptácek L (2004). "Correlating phenotype and genotype in the periodic paralyses". Neurology. 63 (9): 1647–55. doi:10.1212/01.wnl.0000143383.91137.00. PMID 15534250. S2CID 36507153.
- ^ a b Plassart E, Eymard B, Maurs L, Hauw J, Lyon-Caen O, Fardeau M, Fontaine B (1996). "Paramyotonia congenita: genotype to phenotype correlations in two families and report of a new mutation in the sodium channel gene". J Neurol Sci. 142 (1–2): 126–33. doi:10.1016/0022-510X(96)00173-6. PMID 8902732. S2CID 8785846.
- ^ Ptácek L, George A, Griggs R, Tawil R, Kallen R, Barchi R, Robertson M, Leppert M (1991). "Identification of a mutation in the gene causing hyperkalemic periodic paralysis". Cell. 67 (5): 1021–7. doi:10.1016/0092-8674(91)90374-8. PMID 1659948. S2CID 12539865.
- ^ Kim J, Hahn Y, Sohn E, Lee Y, Yun J, Kim J, Chung J (2001). "Phenotypic variation of a Thr704Met mutation in skeletal sodium channel gene in a family with paralysis periodica paramyotonica". J Neurol Neurosurg Psychiatry. 70 (5): 618–23. doi:10.1136/jnnp.70.5.618. PMC 1737343. PMID 11309455.
- ^ Brancati F, Valente E, Davies N, Sarkozy A, Sweeney M, LoMonaco M, Pizzuti A, Hanna M, Dallapiccola B (2003). "Severe infantile hyperkalaemic periodic paralysis and paramyotonia congenita: broadening the clinical spectrum associated with the T704M mutation in SCN4A". J Neurol Neurosurg Psychiatry. 74 (9): 1339–41. doi:10.1136/jnnp.74.9.1339. PMC 1738672. PMID 12933953.
- ^ a b c d Ptáĉek L, Tawil R, Griggs R, Meola G, McManis P, Barohn R, Mendell J, Harris C, Spitzer R, Santiago F (1994). "Sodium channel mutations in acetazolamide-responsive myotonia congenita, paramyotonia congenita, and hyperkalemic periodic paralysis". Neurology. 44 (8): 1500–3. doi:10.1212/wnl.44.8.1500. PMID 8058156. S2CID 28470701.
- ^ a b McClatchey A, McKenna-Yasek D, Cros D, Worthen H, Kuncl R, DeSilva S, Cornblath D, Gusella J, Brown R (1992). "Novel mutations in families with unusual and variable disorders of the skeletal muscle sodium channel". Nat Genet. 2 (2): 148–52. doi:10.1038/ng1092-148. PMID 1338909. S2CID 12492661.
- ^ Bouhours M, Luce S, Sternberg D, Willer J, Fontaine B, Tabti N (2005). "A1152D mutation of the Na+ channel causes paramyotonia congenita and emphasizes the role of DIII/S4-S5 linker in fast inactivation". J Physiol. 565 (Pt 2): 415–27. doi:10.1113/jphysiol.2004.081018. PMC 1464511. PMID 15790667.
- ^ a b McClatchey A, Van den Bergh P, Pericak-Vance M, Raskind W, Verellen C, McKenna-Yasek D, Rao K, Haines J, Bird T, Brown R (1992). "Temperature-sensitive mutations in the III-IV cytoplasmic loop region of the skeletal muscle sodium channel gene in paramyotonia congenita". Cell. 68 (4): 769–74. doi:10.1016/0092-8674(92)90151-2. PMID 1310898. S2CID 31831830.
- ^ Lerche H, Heine R, Pika U, George A, Mitrovic N, Browatzki M, Weiss T, Rivet-Bastide M, Franke C, Lomonaco M (1993). "Human sodium channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III-IV linker". J Physiol. 470: 13–22. doi:10.1113/jphysiol.1993.sp019843. PMC 1143902. PMID 8308722.
- ^ Bouhours M, Sternberg D, Davoine C, Ferrer X, Willer J, Fontaine B, Tabti N (2004). "Functional characterization and cold sensitivity of T1313A, a new mutation of the skeletal muscle sodium channel causing paramyotonia congenita in humans". J Physiol. 554 (Pt 3): 635–47. doi:10.1113/jphysiol.2003.053082. PMC 1664790. PMID 14617673.
- ^ a b c d Ptacek L, Gouw L, Kwieciński H, McManis P, Mendell J, Barohn R, George A, Barchi R, Robertson M, Leppert M (1993). "Sodium channel mutations in paramyotonia congenita and hyperkalemic periodic paralysis". Ann Neurol. 33 (3): 300–7. doi:10.1002/ana.410330312. PMID 8388676. S2CID 33366273.
- ^ Wagner S, Lerche H, Mitrovic N, Heine R, George A, Lehmann-Horn F (1997). "A novel sodium channel mutation causing a hyperkalemic paralytic and paramyotonic syndrome with variable clinical expressivity". Neurology. 49 (4): 1018–25. doi:10.1212/wnl.49.4.1018. PMID 9339683. S2CID 18008683.
- ^ Okuda S, Kanda F, Nishimoto K, Sasaki R, Chihara K (2001). "Hyperkalemic periodic paralysis and paramyotonia congenita--a novel sodium channel mutation". J Neurol. 248 (11): 1003–4. doi:10.1007/s004150170059. PMID 11757950. S2CID 26927085.
- ^ a b Ptácek L, George A, Barchi R, Griggs R, Riggs J, Robertson M, Leppert M (1992). "Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita". Neuron. 8 (5): 891–7. doi:10.1016/0896-6273(92)90203-P. PMID 1316765. S2CID 41160865.
- ^ a b c d Meyer-Kleine C, Otto M, Zoll B, Koch M (1994). "Molecular and genetic characterization of German families with paramyotonia congenita and demonstration of founder effect in the Ravensberg families". Hum Genet. 93 (6): 707–10. doi:10.1007/BF00201577. PMID 8005599. S2CID 39722069.
- ^ Lerche H, Mitrovic N, Dubowitz V, Lehmann-Horn F (1996). "Paramyotonia congenita: the R1448P Na+ channel mutation in adult human skeletal muscle". Ann Neurol. 39 (5): 599–608. doi:10.1002/ana.410390509. PMID 8619545. S2CID 8092621.
- ^ Bendahhou S, Cummins T, Kwiecinski H, Waxman S, Ptácek L (1999). "Characterization of a new sodium channel mutation at arginine 1448 associated with moderate Paramyotonia congenita in humans". J Physiol. 518 (2): 337–44. doi:10.1111/j.1469-7793.1999.0337p.x. PMC 2269438. PMID 10381583.
- ^ Sasaki R, Takano H, Kamakura K, Kaida K, Hirata A, Saito M, Tanaka H, Kuzuhara S, Tsuji S (1999). "A novel mutation in the gene for the adult skeletal muscle sodium channel alpha-subunit (SCN4A) that causes paramyotonia congenita of von Eulenburg". Arch Neurol. 56 (6): 692–6. doi:10.1001/archneur.56.6.692. PMID 10369308.
- ^ a b Lehmann-Horn F, Rüdel R, Ricker K (1993). "Non-dystrophic myotonias and periodic paralyses. A European Neuromuscular Center Workshop held 4–6 October 1992, Ulm, Germany". Neuromuscul Disord. 3 (2): 161–8. doi:10.1016/0960-8966(93)90009-9. PMID 7689382. S2CID 20892960.
- ^ Lehmann-Horn F, Rüdel R, Ricker K, Lorković H, Dengler R, Hopf H (1983). "Two cases of adynamia episodica hereditaria: in vitro investigation of muscle cell membrane and contraction parameters". Muscle Nerve. 6 (2): 113–21. doi:10.1002/mus.880060206. PMID 6304507. S2CID 21594294.
- ^ Fontaine B, Khurana T, Hoffman E, Bruns G, Haines J, Trofatter J, Hanson M, Rich J, McFarlane H, Yasek D (1990). "Hyperkalemic periodic paralysis and the adult muscle sodium channel alpha-subunit gene". Science. 250 (4983): 1000–2. Bibcode:1990Sci...250.1000F. doi:10.1126/science.2173143. PMID 2173143.
- ^ Rojas C, Wang J, Schwartz L, Hoffman E, Powell B, Brown R (1991). "A Met-to-Val mutation in the skeletal muscle Na+ channel alpha-subunit in hyperkalaemic periodic paralysis". Nature. 354 (6352): 387–9. Bibcode:1991Natur.354..387R. doi:10.1038/354387a0. PMID 1659668. S2CID 4372717.
- ^ Heine R, Pika U, Lehmann-Horn F (1993). "A novel SCN4A mutation causing myotonia aggravated by cold and potassium". Hum Mol Genet. 2 (9): 1349–53. doi:10.1093/hmg/2.9.1349. PMID 8242056.
- ^ Kelly P, Yang W, Costigan D, Farrell M, Murphy S, Hardiman O (1997). "Paramyotonia congenita and hyperkalemic periodic paralysis associated with a Met 1592 Val substitution in the skeletal muscle sodium channel alpha subunit--a large kindred with a novel phenotype". Neuromuscul Disord. 7 (2): 105–11. doi:10.1016/S0960-8966(96)00429-4. PMID 9131651. S2CID 1174464.
- ^ Wu F, Gordon E, Hoffman E, Cannon S (2005). "A C-terminal skeletal muscle sodium channel mutation associated with myotonia disrupts fast inactivation". J Physiol. 565 (Pt 2): 371–80. doi:10.1113/jphysiol.2005.082909. PMC 1464529. PMID 15774523.
- ^ Vicart S, Sternberg D, Fontaine B, Meola G (2005). "Human skeletal muscle sodium channelopathies". Neurol Sci. 26 (4): 194–202. doi:10.1007/s10072-005-0461-x. PMID 16193245. S2CID 27141272.
- ^ Subramony S, Malhotra C, Mishra S (1983). "Distinguishing paramyotonia congenita and myotonia congenita by electromyography". Muscle Nerve. 6 (5): 374–9. doi:10.1002/mus.880060506. PMID 6888415. S2CID 20268646.
- ^ Streib E; Lane, Russell J. M.; Turnbull, Douglass M.; Hudgson, Peter; Walton, John; Brumback, Roger A.; Gerst, Jeffery W.; Heckmatt, John Z.; et al. (1984). "Evoked response testing in myotonic syndromes". Muscle Nerve. 7 (7): 590–2. doi:10.1002/mus.880070709. PMID 6544373. S2CID 222015799.
- ^ Taminato T, Mori-Yoshimura M, Miki J, Sasaki R, Satoh N, Oya Y, Nishino I, Takahashi Y (2020) Paramyotonia congenita with persistent distal and facial muscle weakness: A case report with literature review. J Neuromuscul Dis
- ^ Becker PE, Paramyotonia congenita (Eulenberg) in Fortschritte der allgemeinen und klinischen Humangenetik. Thieme, Stuttgart (1970).
- ^ Lee, GM; Kim, JB (June 2011). "Hyperkalemic periodic paralysis and paramyotonia congenita caused by a de novo mutation in the SCN4A gene". Neurology Asia. 16 (2): 163–6.
Notes
[edit]- Lehmann-Horn F, Rüdel R, Ricker K (1993). "Non-dystrophic myotonias and periodic paralyses. A European Neuromuscular Center Workshop held 4–6 October 1992, Ulm, Germany". Neuromuscul Disord. 3 (2): 161–8. doi:10.1016/0960-8966(93)90009-9. PMID 7689382. S2CID 20892960.
- Cannon S (2006). "Pathomechanisms in channelopathies of skeletal muscle and brain". Annu Rev Neurosci. 29: 387–415. doi:10.1146/annurev.neuro.29.051605.112815. PMID 16776591.
Further reading
[edit]- Paramyotonia congenita FAQ at the Periodic Paralysis News Desk. The site also hosts a mailing list for patients with the disorder and medical professionals interested in it.
- Fact page from the Muscular Dystrophy Association