ポリチオフェン
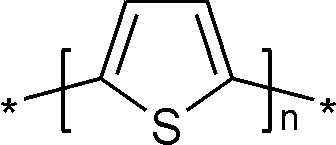

ポリチオフェン (polythiophene, PT) は含硫黄複素環化合物の一種であるチオフェンの重合体(ポリマー)である。ドーピングにより共役π軌道に対して電子を付与または除去すると、導電性を持つようになる。
ポリチオフェン類の研究は1980年代ごろから活発になっていった。導電性ポリマー分野がすでに成熟期を迎えていることは、2000年のノーベル化学賞がアラン・ヒーガー、アラン・マクダイアミッド、そして白川英樹に「導電性ポリマーの発見および発展」における寄与として与えられたことによって確かなものとなった。導電性ポリマーの最も特徴的な性質である電気伝導率は、ポリマー骨格中で電子が非局在化していることによるものである。導電性ポリマーは「合成金属」 (synthetic metals) とも呼ばれる。しかしながら、電子の非局在化によって得られる性質は導電性のみではない。導電性ポリマーは外部からの刺激によって、光学的性質に影響を受ける。すなわち、溶媒・温度・電圧の変化や他の分子との結合により、その色を劇的に変化させる。色と導電性の変化は、共に同じ機構によって起こる。つまりポリマー構造のねじれによって共役系が途切れることに起因する。このような性質を持つことから、導電性ポリマーは光学的・電気的応答を示すセンサーとして魅力あるものとなっている。
ポリチオフェンに関する総説(レビュー)は数多く出版されており、最も初期のものは1981年に発表された[1]。ショップとコスメルは、1990年から1994年にかけて報告された文献の総説を発表している[2]。ロンカリは1992年に電気化学的合成法について[3]、また1997年に置換ポリチオフェン類の電気化学的性質について[4]概説している。マッカローによる1998年の総説は導電性ポリチオフェンの化学合成に焦点を当てたものである[5]。1990年代以降の共役ポリマーに関する総説が、レッディンガーとレイノルズによって1999年に作成されている[6]。そして、スワガーらは共役ポリマーから化学センサーについて2000年にまとめている[7]。これらの総説は、ポリチオフェンに関して、1980年から2000年にかけて報告された主要な一次文献を見出すための助けとなる。
導電性発現の機構とドーピング
[編集]導電性ポリマー中では、電子は共役系にそって非局在化している。これは普通π軌道の重なりを通してのものであり、π系が拡張されて価電子帯を形成している。このπ系から電子を取り除くか(pドーピング)、電子を与えると(nドーピング)、バイポーラロンと呼ばれる電荷を帯びた単位が生成する。

ドーピングは半導体 (<1%) と比べて高い割合 (20%–40%) で施される。バイポーラロンがポリマー鎖の中を移動することによって、巨視的な導電性が発現する。ヨウ素でドーピングしたポリ(3-ドデシル)チオフェンには導電率が 1,000 S/cm に達するものがある[8](銅は約 5× 105 S/cm)。一般的にポリチオフェン類の導電率は 1,000 S/cm を下回るが、導電性ポリマーの用途としては導電性の高さはあまり要求されない。
導電性ポリマーの酸化、および同時に起こる対アニオンの導入(pドーピング)は、電気化学的、もしくは化学的に行うことができる。ポリチオフェンの電気化学的合成を行う場合、ポリマーが電極表面に酸化型となって付着していくのと同時に、溶媒中に溶けている対イオンが取り込まれていく。合成と同時にドーピングを行うことにより、電極上に薄膜が生成していく。この際、生成した導電性ポリマーが基質から膜の表面上に電子を流している。また、中性のポリマー膜、もしくはその溶液を作ったあとでドーピングを行うことも可能である。
導電性ポリマーの還元(nドーピング)はpドーピングと比べ一般的ではない。ポリ(ビチオフェン)の電気化学的nドーピングにおける初期の研究から、nドーピングによるドープ率はpドーピングよりも低いこと、nドーピングのサイクルは効率が悪いこと、ドープ率が最大に達するまでに要するサイクル数が多いこと、そしてnドーピングの過程はおそらく対イオンの分散が原因となって速度論的に制限を受けるようであることが明らかにされている[9]。
ポリチオフェンのドーピングにはさまざまな種類の試薬が使われている。ヨウ素や臭素は導電率の高いものを与えるが[8]、不安定であり、素材中からゆっくりと蒸発していく[10]。トリフルオロ酢酸、プロピオン酸、スルホン酸などの有機酸から得られるポリチオフェンはヨウ素のものより導電率が低いが、外的要因への安定性が高い[10][11]。塩化鉄(III) による酸化重合を行うと、残存する触媒によりドーピングが起こる[12]。ただし、マトリックス支援レーザー脱離イオン化法を使った質量分析 (MALDIMS) による研究から、ポリ(3-ヘキシルチオフェン)について、酸化剤によって部分的にハロゲン化されていることが示されている[13]。トルエン中に溶解したポリ(3-オクチルチオフェン)は塩化鉄(III) 6水和物のアセトニトリル溶液でドーピングすることができ、導電率が 1 S/cm に達する膜として成型することができる[14] 。他、より一般的ではないが、塩化金(III)[15] やトリフルオロメタンスルホン酸[16]もpドーピング剤として知られている。
構造と光学的性質
[編集]共役の長さと色変化
[編集]共役ポリチオフェン類において、π系の拡張は、最も興味が持たれる性質、すなわち光学的性質の元となっている。共役系は近似的に「箱の中の電子」のシュレディンガー方程式の解として考えることができる。しかしながら、一般的なオリゴチオフェン系の吸収および蛍光スペクトルをより正確に予測するため、精密化されたモデルの開発が進められている[17]。共役は芳香環上のπ軌道が重なり合うことによって発生するが、これはすなわち、チオフェン環どうしが同一平面上に存在する必要があることを意味する。
同一平面上にある数が共役系の長さを決定する。そして、共役系が長いほど隣接するエネルギー準位の差は小さくなり、結果として吸収スペクトルに長波長シフトが見られる。平面からのずれの原因には、合成上起こる結合の欠損や、特に立体障害の大きな側鎖による恒久的なものと、外部環境や結合の変化による一時的なものがある。骨格がねじれることによって共役の長さが減少し、エネルギー準位の差が大きくなる。これによって吸収ピークは短波長シフトする。
ポリチオフェンの有効共役長の最大値を決定するためには、立体的に規則ただしい配列と、決まった長さを持つポリチオフェンを合成する必要がある。可視領域の吸収帯は共役が長くなるにつれて長波長シフトするので、最大有効共役長は長波長シフトがそれ以上起こらなくなる点から計算することができる。ヘーヴェらによる初期の研究によると、有効共役長は11個の繰り返し単位にわたって広がっていると見積もられており[18]、一方その後の研究では20単位であるとされている[19]。さらに新しい報告では、大坪らは48[20]および96量体[21]のオリゴチオフェンを合成し、長波長シフトはその変化が小さい(72量体から96量体で 0.1 nm)ものの観測されていることから、有効共役長は96単位よりもさらに長い可能性があるとしている[21]。
共役骨格のねじれ、およびそれに起因する共役長の減少と吸収帯のシフトはさまざまな外的要因によって起こり、溶媒、温度、電圧の印加、溶存イオンがその原因として挙げられる。ポリビニルアルコール水溶液中におけるポリ(3-カルボキシチオフェン)の吸収帯は pH 7 において 480 nm だが、pH 4 では 415 nm にシフトする。この現象は、カルボキシル基が部分的に脱プロトン化することによってポリビニルアルコールとの水素結合を生じるようになり、より圧縮されたコイル型の構造が形成されるためであると考えられている[22] 。キラルなポリチオフェンはクロロホルム中で円偏光二色性を示さないが、クロロホルム/アセトニトリル混合溶媒とクロロホルム/アセトン混合溶媒中ではそれぞれ逆の円偏光二色性を示す[23]。また、キラルなアミノ酸側鎖をもつポリチオフェン[24]はpHと緩衝液の濃度によって吸収帯シフトと円偏光二色性の変化を示す[25]。
温度変化による吸収帯シフトは、低温では平面状・ロッド型の構造をとっているものが、高温ではコイル型に変化するためである。例えば、ポリ(3-オクチルオキシ-4-メチルチオフェン)は25°Cで赤紫色だが150°Cでは淡黄色になる。吸収スペクトル上に等吸収点(吸光度が温度によらず一定である波長)があることから、ふたつの相が存在することが示される(これは同じ鎖の中にあっても、違う鎖の中にあってもよい)[26]。温度による色変化を示すポリチオフェンがすべて等吸収点を持つわけではない。高度なレジオレギュラリティーを持つポリ(3-アルキルチオフェン)類 (PAT) では、低温状態において固体であって結晶状態と不規則相の間の変換が起こらないほど側鎖が十分に短いならば、温度の上昇に伴って短波長シフトが連続的に起こる[要出典]。
吸収帯のシフトは電圧の印加(エレクトロクロミズム)[27]、アルカリ金属イオンの添加(イオノクロミズム)[28]によっても起こすことができる。
3-位置換チオフェンモノマーを2,5-位間で結合させるとき、モノマーの非対称性により3種の構造が考えられる。その構造を以下に示す。
- 2,5', head–tail (HT), カップリング
- 2,2', head–head (HH), カップリング
- 5,5', tail–tail (TT), カップリング
これらの組み合わせにより図に示すような4種の異なる3量体を作ることができる。
それら三量体は核磁気共鳴分光法によって識別することができ、レジオレギュラリティーの度合いを積分によって推定することができる[30][31]。
エルセンバウマーらは、ポリチオフェン類のレジオレギュラリティーの効果に初めて注目した。3-メチルチオフェンおよび3-ブチルチオフェンのレジオランダムの共重合体は50 S/cmの伝導性を有し、さらに、HTとHHカップリングの割合が2:1のレジオレギュラー共重合体は140 S/cmというより大きな伝導性を持つ[32]。94%より多くのHTを含む位置規則性ポリ(3-(4-オクチルフェニル)チオフェン)(POPT)のフィルムは4 S/cmの伝導性を有するのに対し、レジオランダムPOPTでは0.4 S/cmになる[33]。PAT類は、“結晶性、柔軟性そして金属光沢のブロンズ色”の外見の活性亜鉛を使って合成される。一方、類似する位置無作為性ポリマー類は“非結晶質で橙色のフィルム”で合成される[34]。活性PAT類のサーモクロミック特性の比較では、部分規則性ポリマーは強いサーモクロミック効果が見られたが、位置不規則性ポリマーの分光吸光度は高い温度で大きく変化しなかった。これはおそらく局部的な構造欠陥が原因である[要出典]。最後に、スーとホールドクロフトは、HH対の含有量の増加とともにだんだん小さい波長(より大きいエネルギー)で生じるポリ(3-ヘキシルチオフェン)類の蛍光吸収と発光極大を実例で示した。吸収と発光極大との間の差異は、HH対の含有量とともに増加するが、これは最初の励起状態による立体配座の歪みが原因であると考えられる[35]。
溶解度
[編集]非置換のポリチオフェン類はドーピング後伝導性があり、ポリアセチレンのような他の伝導性ポリマーと比べて優れた環境安定性を持つが、加工しにくく、三フッ化ヒ素と五フッ化ヒ素の混合物のような溶媒にしか溶けない[36]。しかしながら、1987年にポリチオフェン類の有機溶媒の例が報告された。エルセンバウマーらはニッケルを触媒としたグリニャールクロスカップリングを使って、ポリ(3-ブチルチオフェン)とポリ(3-メチルチオフェン-'co'-3'-オクチルチオフェン)の2種の溶けるポリチオフェンを合成した。これらをフィルムに成型し、ヨウ素をドープすることによって伝導性が4-6 S/cmに伸ばすことができた[37]。堀田らはポリ(3-ブチルチオフェン)とポリ(3-ヘキシルチオフェン)を合成し、電気化学的[38](と層化学的[39])、そして溶液中でポリマーを特徴付けし[40]、フィルムに成型した[41]。その溶解性のポリチオフェン類は、クロロホルムと2,5-ジメエチルテトラヒドロフラン中でのサーモクロミズムとソルバトクロミズム(先述を参照)が立証された[42]。
また、1987年には、ウドルらによって水溶性のポリ(3-チオフェンアルカンスルホン酸)ナトリウム類の合成法が報告された[43]。水溶性に加えて、スルホン酸側鎖は対イオンとして振る舞い、セルフドープされた伝導性ポリマーを作る。また、カルボン酸[44]、酢酸[45]、アミノ酸[24]およびウレタン類[46]を置換したポリチオフェンも水溶性である。
最近になって、超臨界二酸化炭素中に溶解するポリ(3-(ペルフルオロオクチル)チオフェン)類[47]が、電気化学的、化学的にコラードらによって合成された[48]。
最終的には、熱に不安定なアルキルエステルで両端をキャップされた非置換のオリゴチオフェン類は、溶液からフィルムに成型された後、末端基を水溶化によって除去するために加熱される。加熱の後の原子間力顕微鏡(AFM)画像は、長距離のオーダーに達する大きな増加を示した[49]。
合成
[編集]ポリチオフェン類は、電気化学的にモノマーの溶液に電位を加えるか、化学的に酸化剤もしくはクロスカップリング触媒を使うことによって合成することができるが、それぞれの方法には利点と不利点がある。
電気化学合成
[編集]電気化学的重合では、電位はチオフェンと電解質を含む溶液に適用することにより、陽極上に伝導性ポリチオフェンフィルムを作る[要出典]。電気化学的重合はポリマーを分離・精製する必要が無いため便利であるが、架橋結合のような不規則構造が多かれ少なかれ作られる。
右の図は、モノマーの酸化によってラジカルカチオンが生成し、二番目のラジカルカチオンと繋がってジカチオン二量体を作るか、他のモノマーとラジカルカチオン二量体を作る様子を示したものである。in situビデオ顕微鏡法[50]、サイクリックボルタンメトリー[51]、光電流分光法[52]、電気化学水晶振動子マイクロバランス[53]を含むいくつかのテクニックが、陽極上へのポリマーの沈殿を導く核形成と成長機構の説明に使われた。長時間の沈殿では、電極表面への秩序だったポリマー鎖が、長い/短いフレキシブル鎖になるか、より交差結合された鎖に成長するかは、その重合条件に左右される。
電気化学的に合成されたポリチオフェンフィルムの品質は、いくつかの要因に影響を受ける。その要因には電極の材料、電流密度、温度、溶媒、電解質、水の存在、そしてモノマー濃度が含まれる[2]。この他、モノマーの構造と加電圧も重要であるが、この2つは相互に関係する。モノマーを酸化するための必要な電位は、チオフェン環のπ電子系中の電子密度に依存する。電子供与性基は低酸化電位であり、電子求引性基は高酸化電位である。したがって、アセトニトリルとテトラフルオロホウ酸テトラブチルアンモニウム中で、3-メチルチオフェンと非置換チオフェンをそれぞれ重合させると、前者はSCE(飽和カロメル電極)に対して約1.5 V、非置換チオフェンはSCEに対し約1.7 Vで重合する。しかし、3-置換チオフェンのα炭素に分枝があるとその立体障害により重合が阻害される[54]。したがって、多くのチオフェンモノマーの酸化電位は計算上の酸化電位より大きくなる。これが“ポリチオフェンパラドクス”と呼ばれる所以である。これは、電気化学重合の大きな不利の一つで、複合側基を持つ多くのチオフェンモノマーが制限を受ける。
化学合成
[編集]化学合成は電気化学合成と比較して、モノマーの大きな選択性と適切な触媒を使うことによる完全な規則的置換ポリチオフェン類を合成する能力の2つの点で有利である。ポリチオフェン類は一世紀以上前に偶然化学的に合成されたが[55]、初めて計画的に合成されたのは金属触媒を使った2,5-ジブロモチオフェンの重合で、1980年に二つのグループによって独立に報告された。山本らはテトラヒドロフラン中のマグネシウムとジクロロ(ビピリジン)ニッケルを使ったが、これはハロゲン化アリールとグリニャール試薬の熊田カップリングに類似している[56]。また、リンとドゥーデックはTHF中のマグネシウムとアセチルアセトン錯体の触媒[Pd(acac)2、Ni(acac)2、Co(acac)2、そしてFe(acac)3]を使った[57]。
後の発展でそれら最初の試みより高分子量のポリチオフェン類が合成されるようになったが、その構造に基づき2種に分類することができる。レジオレギュラーポリチオフェン類は、ブロモチオフェンの触媒を用いたクロスカップリング反応によって合成することができる。
完全なレジオレギュラーポリ(3-アルキルチオフェン)類の初合成はマカルーらによって1992年に報告された[58]。右図上のように、選択的臭素化によって2-ブロモ-3-アルキルチオフェンを合成した後、金属交換反応とニッケル触媒による熊田カップリングを行う。NMRスペクトルによると、この方法では約100%のHT-HTカップリングが生成する。その後、右図下のような2,5-ジブロモ-3-アルキルチオフェンを有機金属異性体の混合物を形成する反応性の大きい活性亜鉛[59]で処理する方法がリーケルらによって1993年に報告された[60]。触媒のPd(PPh3)4の量を増やすことで位置無作為性ポリマーが生成するが、Ni(dppe)Cl2で処理することによってレジオレギュラーポリ(3-アルキルチオフェン)が定量的に得られる[61]。
さらに、マカルーとリエケは、低温、無水・無酸素条件、そして臭素化モノマーを必要とする方法で均一な構造のポリ(3-アルキルチオフェン)を合成した。一方、1986年に杉本によって、室温条件で塩化鉄(III)を使ったチオフェンの低負担な酸化重合法が報告された[62]。この方法は、多くの人々によって検証され、H.C. Stark社の帯電防止コーティング材料のバイトロンPは、塩化鉄(III)を使って商用スケールで合成されている[63]。
酸化重合を使って得られる合成物の収率と品質を改善する試みがいくつかの研究で行われている。塩化鉄(III)の他に、塩化鉄(III)水和物、過塩素酸銅そして過塩素酸鉄を含む他の酸化剤もまた2,2'-ビチオフェンの重合によく使われている[64]。モノマー溶液中に塩化鉄(III)をゆっくり加えると、94%のH-Tを含むポリ(3-(4-オクチルフェニル)チオフェン)類が合成される[33]。そこでの塩化鉄(III)の沈殿(触媒の表面積を最大にするため)は、直接モノマーを結晶質の触媒に付加するよりもかなりの高収率でモノマー変換を生成させた[65][66]。重合中に反応混合物を乾燥空気で泡立てることにより、分子量がより大きくなることが報告されている[67]。極性溶媒による重合後の徹底的なソックスレー抽出によって、NMRの測定前にポリマーと触媒の残余が効率的に分画できることが分かっている[30]。モノマーに対し触媒を低比率で用いると(4:1より2:1)、ポリ(3-ドデシルチオフェン)類のレジオレギュラリティーが増加する可能性がある[68]。アンドレアニらは、クロロホルムより四塩化炭素に溶かすことでポリ(ジアルキルチルオフェン)類の高収率を報告したが、これは四塩化炭素中のラジカル種の安定性に起因すると彼らは考えた[69]。高品質触媒を低温低速で加えることによって、残基の無い不溶性ポリマーとともに高分子量のポリ(ジアルキルチルオフェン)類が合成されることが分かっている[70]。ラクソらは、モノマーに対し触媒の比率を高くすることでポリ(3-オクチルチオフェン)の収率が増加することを要因計画を使って究明し、また、長時間の重合も収率を増加させると主張した[71]。
塩化鉄(III)を使った酸化重合の反応機構には議論の余地がある。1986年の杉本らの報告では反応機構の仮説は立てられなかった[62]。しかし、1992年にニエミらによってラジカル機構が提唱された(図のRadicalの部分)。彼らは2つの仮定を反応機構のベースにした。彼らはまず、触媒が難溶または不溶の溶媒(クロロホルム、トルエン、四塩化炭素、ペンタン、ヘキサン)中で重合を観察し、重合の活性点は固体の塩化鉄(III)の表面であることを結論づけた。したがって、彼らは2つのラジカルカチオンが互いに反応する、または2つのラジカルが互いに反応する可能性を軽視した。なぜなら、塩化物イオンが二量化によって活性点だと仮定した結晶の表面でラジカルカチオンもしくはラジカルを抑制するためである[72]。次に彼らは、原型的なモノマーである3-メチルチオフェンを使い、エネルギーと4種の可能な重合種(中性の3-メチルチオフェンとそのラジカルカチオン、2位炭素のラジカルおよび5位炭素のラジカル)の原子荷電の合計を究明するために量子力学的計算を行った。

中性の3-メチルチオフェンの最も陰性な炭素は2位炭素で、ラジカルカチオンの最も奇数電子集団な炭素も2位炭素であるので、ラジカルカチオン機構は主に2-2, H-H結合が優位であると彼らは結論づけた。彼らはそのとき、2位と5位炭素でのラジカル種全ての化学種の全エネルギーを計算し、後者の方が1.5 kJ/molだけ安定なことを発見した。したがって、より安定なラジカルの方が中性種と反応し、H-Tカップリングを形成する(図のRadicalの部分)。
アンダーソンらは、モノマーに触媒をゆっくり混合したときレジオレギュラリティーが高度に見られたため、塩化鉄(III)による3-(4-オクチルフェニル)チオフェンの重合において別の反応機構を提案した。カップリングの選択性を与えると強酸化剤条件では、カルボカチオン機構を通して進むと彼らは結論づけた(図のCarbocationの部分)[33]。
1995年の短報中でラジカル機構は直接的に問題にされ、そのときオリンガとフランソワはチオフェンは、触媒が十分に溶けるアセトニトリル中で塩化鉄(III)によって重合することができたと記述した。また、チオフェン重合の物理的な分析によってもラジカル重合機構の仮定との矛盾が見られた[73]。バーバレラらは3-(アルキルスルファニル)チオフェン類のオリゴマー化を研究し、その量子力学的計算と平面共役オリゴマー上を非局在化したときラジカルカチオンの安定性が改良されたことを考慮した結果、ラジカルカチオン機構は電気化学的重合に類似していると結論づけた(図のRadical cationの部分)[74]。
硬質なロッドポリマーを特徴付けすることを困難にする強酸化剤の触媒の研究、不均一な系の研究の困難さから、酸化重合の機構は明確な意味がない。しかし、ラジカルカチオン機構はポリチオフェン合成のもっとも有望な経路である。
利用
[編集]伝導性ポリチオフェン類の利用が計画されたが、商品化はされなかった。可能性のあるアプリケーションに、電界効果トランジスタ[75]、エレクトロルミネセント素子、太陽電池、フォトレジスト、非線形光学素子[76]、電池、ダイオード、そして化学センサー[77]がある。一般に、伝導性ポリマーのアプリケーションには2つのカテゴリーがある。静的アプリケーションは、高分子材料に共通する加工の容易さと材料特性に加え、材料の真性導電率を活用するものである。動的アプリケーションでは、電位の負荷または環境刺激から生じる導電性と光学特性の変化が利用される。

静的アプリケーションの例に、帯電(静電気)防止塗料であるH.C. Starck社のポリ(3,4-エチレンジオキシチオフェン)-ポリ(スチレンスルホン酸)(PEDOT-PSS、商品名:バイトロンP)がある。アグフア・ゲバルト社はその帯電防止特性のため、バイトロンとともに年に200 m×10 mの写真用フィルムを被覆している。バイトロンの薄層は無色透明で、フィルム巻き戻し中ぎ静電放電を防ぎ、処理後のネガ上へのホコリの付着を減らす。
また、ポリ(3,4-エチレンジオキシチオフェン)(PEDOT)は動的アプリケーションに使うこともできる。PEDOTのエレクトロクロミズムの性質は、電位の適用によって不透明になったり反射状態になったりする窓や鏡の製造に使われている[27]。エレクトロクロミックウインドウの広範囲に渡る採用により空調のコストを大幅に節約することができている[78]。
また、検体に反応するセンサーとしてのポリチオフェン類の用途は広く研究の対象となっている。さらに、バイオセンサーへの利用に加えて、ポリチオフェン類は金属イオンまたはキラルな分子を検出するための合成受容体として官能基化することもできる。側鎖[79]と主鎖[28]がクラウンエーテル化されたポリチオフェンがBäuerleとSwagerの研究グループによって1993年にそれぞれ報告された。電気化学重合されたBäuerle側鎖クラウンエーテルポリチオフェンの薄膜は、ミリモーラーの濃度のアルカリカチオン(Li, Na, K)に晒された。カリウムイオン溶液では僅かに、ナトリウム溶液では少し、リチウムイオン溶液中では著しく落ちた固定電位でフィルムを電流が流れた。一方、Swager主鎖クラウンエーテルポリチオフェンは化学合成で作られ、紫外可視吸収スペクトルで特徴付けされた。加えて、アルカリカチオンでの吸光度シフトは46 nm (Li) 、91 nm (Na)、22 nm (K)という結果になった。シフトの大きさは、対応するクラウンエーテルのイオン捕捉に対応し、イオンの捕捉によるポリマー骨格のねじれが原因である。
キラルポリチオフェン類の光学的性質が研究されていた間[80][81][82][83]、八島と五藤は、キラルなアミノアルコールを感知し、π-遷移領域に鏡像分割誘起円二色性を作るキラルな一級アミンのついたポリチオフェンを報告した[84]。これは、キラル検出法(円偏光二色性)にポリチオフェン類を使った初の不斉認識の例である。
参考文献
[編集]- Handbook of Conducting Polymers (Eds: T. A. Skotheim, R. L. Elsenbaumer, J. R. Reynolds), Marcel Dekker, New York 1998. ISBN 0-8247-0050-3.
- G. Schopf, G. Koßmehl, Polythiophenes: Electrically Conductive Polymers, Springer, Berlin 1997. ISBN 3-540-61483-4; ISBN 0-387-61483-4.
- Synthetic Metals (journal). ISSN 0379-6779.
脚注
[編集]- ^ Street, G. B.; Clarke, T. C. IBM J. Res. Dev. 1981, 25, 51–57.
- ^ a b Schopf, G.; Koßmehl, G. Adv. Polym. Sci. 1997, 129, 1–166.
- ^ Roncali, J. Chem. Rev. 1992, 92, 711–738. doi:10.1021/cr00012a009.
- ^ Roncali, J. Chem. Rev. 1997, 97, 173–205. doi:10.1021/cr950257t.
- ^ McCullough, R. D. Adv. Mater. 1998, 10, 93–116.
- ^ Reddinger, J. L.; Reynolds, J. R. Adv. Polym. Sci. 1999, 145, 57–122.
- ^ McQuade, D. T.; Pullen, A. E.; Swager, T. M. Chem. Rev. 2000, 100, 2537–2574. doi:10.1021/cr9801014.
- ^ a b McCullough, R. D.; Tristramnagle, S.; Williams, S. P.; Lowe, R. D.; Jayaraman, M. J. Am. Chem. Soc. 1993, 115, 4910–4911. doi:10.1021/ja00064a070.
- ^ Mastragostino, M.; Soddu, L. Electrochim. Acta 1990, 35, 463–466. doi:10.1016/0013-4686(90)87029-2.
- ^ a b Loponen, M. T.; Taka, T.; Laakso, J.; Väkiparta, K.; Suuronen, K.; Valkeinen, P.; Österholm, J. E. Synth. Met. 1991, 41, 479–484. doi:10.1016/0379-6779(91)91111-M.
- ^ Bartus, J. J. Macromol. Sci., Chem. 1991, A28, 917–924.
- ^ Qiao, X. Y.; Wang, X. H.; Mo, Z. S. Synth. Met. 2001, 122, 449–454.
- ^ McCarley, T. D.; Noble, C. O.; DuBois, C. J.; McCarley, R. L. Macromolecules 2001, 34, 7999–8004. doi:10.1021/ma002140z.
- ^ Heffner, G. W.; Pearson, D. S. Synth. Met. 1991, 44, 341–347. doi:10.1016/0379-6779(91)91821-Q.
- ^ Abdou, M. S. A.; Holdcroft, S. Synth. Met. 1993, 60, 93–96. doi:10.1016/0379-6779(93)91226-R.
- ^ Rudge, A.; Raistrick, I.; Gottesfeld, S.; Ferraris, J. P. Electrochim. Acta 1994, 39, 273–287. doi:10.1016/0013-4686(94)80063-4.
- ^ Bässler, H. Electronic Excitation. In Electronic Materials: The Oligomer Approach; Müllen, K.; Wegner, G., Eds.; Wiley-VCH: Weinheim, Germany 1998. ISBN 3-527-29438-4
- ^ ten Hoeve, W.; Wynberg, H.; Havinga, E. E.; Meijer, E. W. J. Am. Chem. Soc. 1991, 113, 5887–5889. doi:10.1021/ja00015a067
- ^ Meier, H.; Stalmach, U.; Kolshorn, H. Acta Polym., 48, 379–384.
- ^ Nakanishi, H.; Sumi, N.; Aso, Y.; Otsubo, T. J. Org. Chem. 1998, 63, 8632–8633. doi:10.1021/jo981541y
- ^ a b Izumi, T.; Kobashi, S.; Takimiya, K.; Aso, Y.; Otsubo, T. J. Am. Chem. Soc. 2003, 125, 5286–5287. doi:10.1021/ja034333i
- ^ de Souza, J. M.; Pereira, E. C. Synth. Met. 2001, 118, 167–170. doi:10.1016/S0379-6779(00)00453-7
- ^ Goto, H.; Yashima, E.; Okamoto, Y. Chirality 2000, 12, 396–399.
- ^ a b Andersson, M.; Ekeblad, P. O.; Hjertberg, T.; Wennerström, O.; Inganäs, O. Polym. Commun. 1991, 32, 546–548.
- ^ Nilsson, K. P. R.; Andersson, M. R.; Inganäs, O. J. Phys.: Condes. Matter 2002, 14, 10011–10020.
- ^ Roux, C.; Leclerc, M. Macromolecules 1992, 25, 2141–2144. doi:10.1021/ma00034a012
- ^ a b Heuer, H. W.; Wehrmann, R.; Kirchmeyer, S. Adv. Funct. Mater. 2002, 12, 89–94. doi:10.1002/1616-3028(20020201)12:2<89::AID-ADFM89>3.0.CO;2-1
- ^ a b Marsella, M. J.; Swager, T. M. J. Am. Chem. Soc. 1993, 115, 12214–12215. doi:10.1021/ja00078a090
- ^ 正確には高分子用語として主鎖に不斉点を持たないポリチオフェンに立体規則性は存在しないが、用語の誤用が広まっているため、ポリチオフェンに限りレジオレギュラリティーのことを立体規則性と表記することがある。
- ^ a b Barbarella, G.; Bongini, A.; Zambianchi, M. Macromolecules 1994, 27, 3039–3045. doi:10.1021/ma00089a022
- ^ Diaz-Quijada, G. A.; Pinto, B. M.; Holdcroft, S. Macromolecules 1996, 29, 5416–5421. doi:10.1021/ma960126+
- ^ Elsenbaumer, R. L.; Jen, K.-Y.; Miller, G. G.; Eckhardt, H.; Shacklette, L. W.; Jow, R. "Poly (alkyl thiophenes) and Poly (substituted heteroaromatic vinylenes): Versatile, Highly Conductive, Processible Polymers with Tunable Properties." In Electronic Properties of Conjugated Polymers (Eds: Kuzmany, H.; Mehring, M.; Roth, S.), Springer, Berlin, 1987. ISBN 0-387-18582-8
- ^ a b c Andersson, M. R.; Selse, D.; Berggren, M.; Järvinen, H.; Hjertberg, T.; Inganäs, O.; Wennerström, O.; Österholm, J. E. Macromolecules 1994, 27, 6503–6506. doi:10.1021/ma00100a039
- ^ Chen, T. A.; Wu, X.; Rieke, R. D. J. Am. Chem. Soc. 1995, 117, 233–244. doi:10.1021/ja00106a027
- ^ Xu, B.; Holdcroft, S. Macromolecules 1993, 26, 4457–4460. doi:10.1021/ma00069a009
- ^ Frommer, J. E. Acc. Chem. Res. 1986, 19, 2–9. doi:10.1021/ar00121a001
- ^ Elsenbaumer, R. L.; Jen, K. Y.; Oboodi, R. Synth. Met. 1986, 15, 169–174. doi:10.1016/0379-6779(86)90020-2
- ^ Hotta, S.; Rughooputh, S. D. D. V.; Heeger, A. J.; Wudl, F. Macromolecules 1987, 20, 212–215. doi:10.1021/ma00167a038
- ^ Hotta, S.; Soga, M.; Sonoda, N. Synth. Met. 1988, 26, 267–279. doi:10.1016/0379-6779(88)90243-3
- ^ Hotta, S. Synth. Met. 1987, 22, 103–113. doi:10.1016/0379-6779(87)90528-5
- ^ Hotta, S.; Rughooputh, S. D. D. V.; Heeger, A. J. Synth. Met. 1987, 22, 79–87. doi:10.1016/0379-6779(87)90573-X
- ^ Rughooputh, S. D. D. V.; Hotta, S.; Heeger, A. J.; Wudl, F. J. Polym. Sci., Polym. Phys. Ed. 1987, 25, 1071–1078.
- ^ Patil, A. O.; Ikenoue, Y.; Wudl, F.; Heeger, A. J. J. Am. Chem. Soc. 1987, 109, 1858–1859. doi:10.1021/ja00240a044
- ^ Englebienne, P.; Weiland, M. Chem. Commun. 1996, 1651–1652. doi:10.1039/cc9960001651
- ^ Kim, B. S.; Chen, L.; Gong, J. P.; Osada, Y. Macromolecules 1999, 32, 3964–3969. doi:10.1021/ma981848z
- ^ Jung, S. D.; Hwang, D. H.; Zyung, T.; Kim, W. H.; Chittibabu, K. G.; Tripathy, S. K. Synth. Met. 1998, 98, 107–111.
- ^ DeSimone, J. M.; Guan, Z.; Elsbernd, C. S. Science 1992, 257, 945–947.
- ^ Li, L.; Counts, K. E.; Kurosawa, S.; Teja, A. S.; Collard, D. M. Adv. Mater. 2004, 16, 180–183.
- ^ Murphy, A. R.; Fréchet, J. M. J.; Chang, P.; Lee, J.; Subramanian, V. J. Am. Chem. Soc. 2004, 126, 1596–1597.
- ^ Lukkari, J.; Tuomala, R.; Ristimaki, S.; Kankare, J. Synth. Met. 1992, 47, 217–231.
- ^ Lukkari, J.; Kankare, J.; Visy, C. Synth. Met. 1992, 48, 181–192.
- ^ Lukkari, J.; Alanko, M.; Pitkänen, V.; Kleemola, K.; Kankare, J. J. Phys. Chem. 1994, 98, 8525–8535.
- ^ Visy, C.; Lukkari, J.; Kankare, J. Synth. Met. 1997, 87, 81–87.
- ^ Roncali, J.; Garreau, R.; Yassar, A.; Marque, P.; Garnier, F.; Lemaire, M. J. Phys. Chem. 1987, 91, 6706–6714.
- ^ Meyer, V. Ber. Deutsch. Chem. Ges. 1883, 16, 1465–1478.
- ^ Yamamoto, T.; Sanechika, K.; Yamamoto, A. J. Polym. Sci., Polym. Lett. Ed. 1980, 18, 9–12.
- ^ Lin, J. W. P.; Dudek, L. P. J. Polym. Sci., Polym. Chem. Ed. 1980, 18, 2869–2873.
- ^ McCullough, R. D.; Lowe, R. D. J. Chem. Soc., Chem. Commun. 1992, 70–72.
- ^ Zhu, L.; Wehmeyer, R. M.; Rieke, R. D. J. Org. Chem. 1991, 56, 1445–1453.
- ^ Chen, T. A.; O’Brien, R. A.; Rieke, R. D. Macromolecules 1993, 26, 3462–3463.
- ^ Chen, T. A.; Rieke, R. D. J. Am. Chem. Soc. 1992, 114, 10087–10088.
- ^ a b Sugimoto, R.; Taketa, S.; Gu, H. B.; Yoshino, K. Chem. Express 1986, 1, 635–638.
- ^ Jonas, F.; Heywang, G.; Schmidtberg, W.; Heinze, J.; Dietrich, M. アメリカ合衆国特許第 5,035,926号, 1991.
- ^ Ruckenstein, E.; Park, J. S. Synth. Met. 1991, 44, 293–306.
- ^ Bizzarri, P. C.; Andreani, F.; Della Casa, C.; Lanzi, M.; Salatelli, E. Synth. Met. 1995, 75, 141–147.
- ^ Fraleoni-Morgera, A.; Della Casa, C.; Lanzi, M.; Bizzarri, P. C. Macromolecules 2003, 36, 8617–8620. doi:10.1021/ma0348730
- ^ Pomerantz, M.; Tseng, J. J.; Zhu, H.; Sproull, S. J.; Reynolds, J. R.; Uitz, R.; Arnott, H. J.; Haider, M. I. Synth. Met. 1991, 41, 825–830.
- ^ Qiao, X. Y.; Wang, X. H.; Zhao, X. J.; Liu, J.; Mo, Z. S. Synth. Met. 2000, 114, 261–265.
- ^ Andreani, F.; Salatelli, E.; Lanzi, M. Polymer 1996, 37, 661–665.
- ^ Gallazzi, M. C.; Bertarelli, C.; Montoneri, E. Synth. Met. 2002, 128, 91–95.
- ^ Laakso, J.; Järvinen, H.; Skagerberg, B. Synth. Met. 1993, 55, 1204–1208.
- ^ Niemi, V. M.; Knuuttila, P.; Österholm, J. E.; Korvola, J. Polymer 1992, 33, 1559–1562.
- ^ Olinga, T.; François, B. Synth. Met. 1995, 69, 297–298.
- ^ Barbarella, G.; Zambianchi, M.; DiToro, R.; Colonna, M.; Iarossi, D.; Goldoni, F.; Bongini, A. J. Org. Chem. 1996, 61, 8285–8292.
- ^ Garnier, F. Field-Effect Transistors Based on Conjugated Materials. In Electronic Materials: The Oligomer Approach (Eds: Müllen, K.; Wegner, G.), Wiley-VCH, Weinheim, 1998. ISBN 3-527-29438-4
- ^ Harrison, M. G.; Friend, R. H. Optical Applications. In Electronic Materials: The Oligomer Approach (Eds: Müllen, K.; Wegner, G.), Wiley-VCH, Weinheim, 1998. ISBN 3-527-29438-4
- ^ Martina, V.; Ionescu, K.; Pigani, L.; Terzi, F.; Ulrici, A.; Zanardi, C.; Seeber, R. Anal. Bioanal. Chem. 2007, 387, 2101-2110.
- ^ Rosseinsky, D. R.; Mortimer, R. J. Adv. Mater. 2001, 13, 783–793.
- ^ Bäuerle, P.; Scheib, S. Adv. Mater. 1993, 5, 848–853.
- ^ Goto, H.; Okamoto, Y.; Yashima, E. Chem. Eur. J. 2002, 8, 4027–4036.
- ^ Goto, H.; Okamoto, Y.; Yashima, E. Macromolecules 2002, 35, 4590–4601.
- ^ Goto, H.; Yashima, E. J. Am. Chem. Soc. 2002, 124, 7943–7949.
- ^ Sakurai, S.; Goto, H.; Yashima, E. Org. Lett. 2001, 3, 2379–2382.
- ^ Yashima, E.; Goto, H.; Okamoto, Y. Macromolecules 1999, 32, 7942–7945.