Inhibidor encimático
Os inhibidores encimáticos son moléculas que se unen a encimas e diminúen a súa actividade. Como o bloqueo dun encima pode matar a un organismo patóxeno ou corrixir un desequilibrio metabólico, moitos medicamentos actúan como inhibidores encimáticos. Tamén se usan como herbicidas e pesticidas. Porén, non todas as moléculas que se unen a encimas son inhibidores; existen os activadores encimáticos, que se unen a encimas incrementando a súa actividade.
A unión dun inhibidor pode impedir a entrada do substrato ao sitio activo do encima e/ou empecer que o encima catalice a súa reacción correspondente. A unión do inhibidor pode ser reversible ou irreversible. Normalmente, os inhibidores irreversibles reaccionan co encima de forma covalente e modifican a súa estrutura química en residuos esenciais dos aminoácidos necesarios para a actividade encimática. Polo contrario, os inhibidores reversibles únense ao encima de forma non covalente, dando lugar a diferentes tipos de inhibicións, dependendo de se o inhibidor se une ao encima, ao complexo encima-substrato ou a ambos os dous.
Moitos medicamentos son inhibidores encimáticos, polo que o seu descubrimento e mellora é un eido de investigación moi activo en bioquímica e farmacoloxía. A validez dun inhibidor encimático medicinal adoita vir determinada pola súa especificidade (a súa incapacidade de unirse a outras proteínas) e a súa potencia (a súa constante de disociación, a cal indica a concentración necesaria para inhibir un encima). Unha alta especificidade e potencia asegura que o medicamento vai ter poucos efectos secundarios e, por tanto, unha baixa toxicidade.
Os inhibidores encimáticos tamén se usan na natureza e están implicados na regulación do metabolismo. Por exemplo, os encimas dunha ruta metabólica poden ser inhibidos polos produtos resultantes das súas respectivas rutas. Este tipo de retroalimentación negativa retarda o fluxo a través da ruta cando os produtos comezan a acumularse e é unha maneira importante de manter a homeostase nunha célula. Outros inhibidores encimáticos celulares son proteínas que se unen especificamente e inhiben unha diana encimática. Isto pode axudar a controlar encimas que poden ser daniños para a célula, como as proteases ou nucleases. Un bo exemplo é o inhibidor da ribonuclease, que se une a este encima nunha das interaccións proteína-proteína máis fortes coñecidas.[1] Como inhibidores encimáticos naturais tamén hai que salientar os velenos, que son usados como defensa contra os depredadores ou como forma de matar a unha presa.
Inhibidores reversibles
editarOs inhibidores reversibles únense aos encimas por medio de interaccións non covalentes tales como as pontes de hidróxeno, as interaccións hidrofóbicas e os enlaces iónicos. Os enlaces febles múltiples entre o inhibidor e o sitio activo combínanse para producir unha unión forte e específica. Ao contrario do que ocorre co substrato e os inhibidores irreversibles, os inhibidores reversibles xeralmente non experimentan reaccións químicas cando se unen ao encima e poden ser eliminados doadamente por dilución ou por diálise.
Tipos de inhibidores reversibles
editarExisten tres tipos de inhibidores reversibles. Clasifícanse baseándose no efecto producido pola variación da concentración do substrato do encima no inhibidor.[2]
- Na inhibición competitiva, o substrato e o inhibidor non se poden unir ao mesmo encima ao mesmo tempo, como se mostra na figura da dereita. Isto xeralmente ocorre porque o inhibidor e o substrato teñen unha estrutura similar e ambos os dous teñen afinidade polo sitio activo, e o substrato e o inhibidor compiten para entrar nel. Este tipo de inhibición pódese superar con concentracións suficientemente altas de substrato, que deixan fóra de competición ao inhibidor. O cambio nas concentracións de inhibidor fan cambiar a Km pero non a Vmax[2]
- A inhibición acompetitiva ou incompetitiva (uncompetitive inhibition) prodúcese cando o inhibidor se une só ao complexo encima-substrato, non ao encima libre, é dicir, se non está unido o substrato o inhibidor non se pode unir. O complexo EIS é cataliticamente inactivo. Esta forma de inhibición é rara e causa unha diminución tanto no valor de Vmax coma no de Km.[3] Non hai que confundila coa inhibición non-competitiva (non-competitive inhibition).
- Na inhibición mixta, o inhibidor pódese unir ao encima ao mesmo tempo que o substrato ou por separado. Porén, a unión do inhibidor afecta á unión do substrato, e viceversa. Este tipo de inhibición pódese reducir, pero non superar aumentando as concentracións do substrato. Aínda que é posible que os inhibidores de tipo mixto se unan no sitio activo, este tipo de inhibición orixínase xeralmente por un efecto alostérico onde o inhibidor se une a outro sitio distinto do sitio activo do encima. A unión do inhibidor co sitio alostérico fai mudar a conformación (é dicir, a estrutura terciaria ou a forma tridimensional) do encima de tal xeito que a afinidade do substrato polo sitio activo se reduce.[2] Varía tanto a Km coma a Vmax.
- A inhibición non-competitiva é unha forma de inhibición mixta onde a unión do inhibidor co encima reduce a súa actividade pero non afecta á unión co substrato. Como resultado, o grao de inhibición depende soamente da concentración de inhibidor.[2] Varía a Vmax pero non a Km. O inhibidor ten a mesma afinidade de unión co encima tanto se este está unido ao substrato coma se non.
Descrición cuantitativa da inhibición reversible
editarA inhibición reversible pode ser descrita cuantitativamente en termos da unión do inhibidor ao encima e ao complexo encima-substrato, e os seus efectos nas constantes cinéticas do encima. No esquema clásico de Michaelis-Menten mostrado abaixo, un encima (E) únese ao seu substrato (S) para formar o complexo encima-substrato (ES). Na catálise, este complexo rompe para liberar o produto (P) e o encima (E). O inhibidor (I) pode unirse tanto a (E) coma a (ES) coas constantes de disociación Ki ou Ki', respectivamente.
|
Cando un encima ten múltiples substratos, os inhibidores poden mostrar distintos tipos de inhibicións dependendo do substrato que se considere, xa que o sitio activo posúe dous diferentes lugares para a unión co substrato no mesmo sitio activo, un para cada substrato. Por exemplo, un inhibidor pode competir co substrato A polo primeiro sitio de unión, pero ser un inhibidor non competitivo con respecto ao substrato B no segundo sitio de unión.[3]
Medición das constantes de disociación nun inhibidor reversible
editarSegundo o observado arriba, un inhibidor encimático está caracterizado polas súas dúas constantes de disociación, Ki e Ki', do encima e do complexo encima-substrato, respectivamente. A constante do complexo encima-inhibidor Ki pode ser medida directamente por varios métodos. Un método estremadamente exacto é a calorimetría isoterma de titulación, en onde o inhibidor é titulado nunha solución de encimas e mídese a calor liberada ou absorbida.[4] Porén, a outra constante de disociación Ki' é difícil de medir directamente, xa que o complexo encima-substrato ten un período de vida moi curto e está dando lugar á reacción química para formar o produto. Polo tanto, Ki' adoita medirse de forma indirecta, observando a actividade encimática baixo varias concentracións de substrato e inhibidor, e axustando os datos [5] a unha ecuación de Michaelis-Menten modificada:
onde os factores de modificación α e α' son definidos pola concentración do inhibidor e as súas dúas constantes de disociación
Así, en presenza do inhibidor, a efectividade do encima Km e Vmax é agora (α/α')Km e (1/α')Vmax, respectivamente. Porén, a ecuación de Michaelis-Menten modificada asume que a unión do inhibidor ao encima atingue o equilibrio, o cal pode ser un proceso moi lento para os inhibidores con constantes secundarias nanomolares de disociación. Nestes casos, é usualmente máis práctico tratar ao inhibidor de unión forte como un inhibidor irreversible (ver abaixo). Porén, aínda pode ser posible estimar Ki' cineticamente se Ki se mide independentemente.[5]
Os efectos de diferentes tipos de inhibidores encimáticos reversibles na actividade encimática poden ser visualizados usando a representación gráfica da ecuación de Michaelis-Menten, por medio dos diagramas de Lineweaver-Burk ou de Eadie-Hofstee. Por exemplo, nos diagramas de Lineweaver-Burk á dereita, as liñas da inhibición competitiva cortan o eixe-y, o que indica que tales inhibidores non afectan á Vmax. De igual maneira, as liñas da inhibición non-competitiva cortan o eixe-x, mostrando que estes inhibidores non afectan á Km. Porén, pode ser complicado estimar Ki e Ki' con precisión nestes diagramas, polo que é recomendable estimar estas constantes usando métodos máis fiables de regresión non liñal,[6] segundo o descrito arriba.
Casos especiais
editar- O mecanismo da inhibición parcialmente competitiva é similar ao da inhibición non-competitiva, excepto que o complexo EIS ten actividade catalítica, a cal decrece ou mesmo aumenta (activación parcialmente competitiva) en comparación co complexo encima-substrato (ES). Esta inhibición adoita ter un valor máis baixo de Vmax, pero un valor de Km inalterado.[3]
- A inhibición por substrato e por produto é aquela na que o substrato ou o produto dunha reacción encimática inhiben a actividade encimática. Este tipo de inhibición pode seguir os patróns competitivos, non competitivos ou mixtos. Na inhibición por substrato hai unha diminución progresiva da actividade a altas concentracións de substrato. Isto pode indicar a existencia de dous sitios de unión entre substrato e encima. Cando hai pouco substrato, ocúpase o sitio de alta afinidade e segue a cinética normal. Porén, a altas concentracións, ocúpase o segundo sitio de inhibición, inhibindo ao encima.[7] A inhibición por parte do produto é a miúdo unha característica reguladora no metabolismo e pode ser unha forma de retroalimentación negativa.
- A inhibición lenta e forte prodúcese cando o complexo encima-inhibidor EI inicial experimenta unha isomerización a un segundo complexo máis fortemente unido, EI*, pero o proceso total da inhibición é reversible. Isto maniféstase como un lento aumento na inhibición encimática. Nestas condicións, a tradicional cinética de Michaelis–Menten dá un valor falso para Ki, o cal depende do tempo. O verdadeiro valor de Ki pode ser obtido a través dunha análise máis complexa das constantes de rango de encendido (kon) e apagado (koff) para a asociación do inhibidor (véxase a inhibición irreversible para máis información).[3][7]
Exemplos de inhibidores reversibles
editarComo os encimas evolucionaron para unirse aos seus substratos fortemente, e a maioría dos inhibidores reversibles se unen ao sitio activo dos encimas, é pouco sorprendente que algúns destes inhibidores sexan moi similares en estrutura aos substratos das súas dianas. Como exemplo destes imitadores de substratos salientaremos os inhibidores da protease, unha clase moi efectiva de fármacos antirretrovirais usados para tratar a infección por VIH. A estrutura do ritonavir (figura da dereita), un inhibidor da protease, consiste nun péptido con tres enlaces peptídicos. Dita estrutura aseméllase á proteína que é o substrato da protease do VIH, polo que ambos os dous compiten pola unión ao sitio activo do encima.[8]
Os inhibidores encimáticos son a miúdo deseñados para imitaren o estado de transición ou intermedio dunha reacción catalizada por un encima. Isto asegura que o inhibidor cambie o estado de transición establecendo un efecto no encima, o que dá lugar a unha mellor afinidade na unión (baixa Ki) cós deseños baseados en substratos. Un exemplo dun inhibidor nese estado de transición é o fármaco antiviral oseltamivir, que imita a natureza plana do anel do ión oxonio na reacción da neuraminidase, un encima do virus.[9]
Porén, non todos os inhibidores están baseados na estrutura do substrato. Por exemplo, a estrutura doutro inhibidor da protease do VIH, o tipranavir, (representada á dereita), non está baseada nun péptido e non ten similitudes estruturais evidentes coa proteína substrato. Estes inhibidores non peptídicos poden ser máis estables cós inhibidores que conteñen enlaces peptídicos porque estes non son substratos para as peptidases, co que son menos propensos á degradación na célula.[10]
No deseño de fármacos é importante considerar as concentracións de substrato ás cales se expoñerá o encima en cuestión. Por exemplo, algúns inhibidores de proteín-quinases teñen estruturas químicas que son similares ao adenosín trifosfato, un dos substratos deste encima. Porén, certos fármacos que son simplemente inhibidores competitivos terán que competir con altas concentracións de ATP na célula. As proteín-quinases tamén poden ser inhibidas por competencia no sitio de unión onde a quinase interacciona coas súas proteínas substrato, e a maioría das proteínas presentes no interior dunha célula encóntranse a concentracións moito menores cás concentracións de ATP. En consecuencia, se dous inhibidores de proteín-quinases se unen nos seus sitios activos con afinidade similar, pero só un ten que competir co ATP, entón o inhibidor competitivo no sitio de unión da proteína inhibirá ao encima máis eficientemente.[11]
Inhibidores irreversibles
editarOs inhibidores irreversibles normalmente modifican un encima covalentemente, co cal a inhibición non pode inverterse. Os inhibidores irreversibles adoitan conter grupos funcionais reactivos como mostazas nitroxenadas, aldehidos, haloalcanos ou alquenos. Estes grupos electrofílicos reaccionan coas cadeas de aminoácidos para formar unións covalentes. Os residuos modificados son aqueles que conteñen nas súas cadeas laterais nucleófilos como por exemplo un grupo hidroxilo ou un grupo sulfhidrilo. Isto inclúe aos aminoácidos serina (como no DFP, á dereita), cisteína, treonina ou tirosina.[12]
Tipos de inhibiciones irreversibles
editarA inhibición irreversible é diferente da inactivación encimática reversible. Os inhibidores irreversibles son xeralmente específicos para un tipo de encima e non inactivan a todas as proteínas. Non funcionan destruíndo os enlaces peptídicos da estrutura proteínica, senón alterando especificamente a estrutura tridimensional do sitio activo inhabilitándoo. Por exemplo, o pH e as temperaturas estremas causan a desnaturalización de case todas as proteínas, pero este non é un efecto específico. De forma similar, algúns tratamentos químicos non específicos destrúen a estrutura de enlaces peptídicos da proteína: por exemplo, se son sometidas a unha elevada concentración de ácido clorhídrico, o cal hidrolizará os enlaces peptídicos que manteñen unidos os aminoácidos das proteínas.[13]
Os inhibidores irreversibles dan lugar a unha inhibición dependente do tempo e, por iso, a súa potencia non pode ser caracterizada por medio da determinación do valor IC50. Isto débese a que a cantidade de encima activa a unha concentración dada de inhibidor irreversible será diferente dependendo do tempo de pre-incubación do inhibidor co encima. Por tanto, en lugar do valor IC50, utilízase o parámetro kobs/[I],[14] no que kobs é o primeiro valor observado da taxa de inactivación (obtido ao representarmos nunha gráfica log %actividade VS. tempo) e [I] é a concentración de inhibidor. O parámetro kobs/[I] é válido sempre e cando o inhibidor non se encontre a concentracións saturantes (nese caso teríamos que kobs = kinact).[14]
Análise da inhibición irreversible
editarComo se mostra na figura da esquerda, os inhibidores irreversibles forman inicialmente un complexo reversible e non covalente co encima (EI ou ESI), que reaccionará posteriormente para producir unha modificación covalente no que se denomina o "complexo do punto morto" EI*. A taxa á cal se forma EI* chámase taxa de inactivación ou kinact. Dado que a formación de EI pode competir con ES, a unión dos inhibidores irreversibles pode previrse por competencia tanto co substrato coma cun segundo inhibidor reversible. Este efecto de protección é unha boa evidencia dunha reacción específica do inhibidor irreversible co sitio activo.[15]
Os pasos de unión e inactivación desta reacción analízanse incubando o encima co inhibidor e medindo a actividade que vai quedando ao longo do tempo. A actividade irá diminuíndo de forma paulatina co tempo, xeralmente seguindo unha dinámica de decaimento expoñencial. Axustando estes datos a unha ecuación de rango podemos obter a taxa de inactivación a esta concentración de inhibidor. Isto faise a diferentes concentracións de inhibidor. Se está implicado un complexo EI reversible o rango de inactivación será saturable, e ao axustala a esta curva obteremos os valores de kinact y Ki.[15]
Outro método que se utiliza moito nestas análises é a espectrometría de masas. Neste caso, a medida exacta da masa do encima nativo sen modificar e do encima inactivado, dános o aumento na masa causado pola reacción co inhibidor, co que obtemos a estequiometría da reacción. Para levar a cabo esta técnica adoita ser necesario o uso dun espectrómetro de masas MALDI-TOF. Unha técnica complementaria a esta é a pegada peptídica, que implica a dixestión de proteínas nativas ou modificadas cunha peptidase como a tripsina. Isto xera unha serie de péptidos que poden analizarse usando un espectrómetro de masas. O péptido que cambie a súa masa despois de levar a cabo a reacción co inhibidor será aquel que conteña o sitio da modificación.[16]
Casos especiais
editarNon todos os inhibidores irreversibles forman unións covalentes co encima que teñen como diana. Algúns inhibidores reversibles únense tan fortemente ao seu obxectivo que son practicamente irreversibles. Estes inhibidores de unión forte mostran xeralmente unha cinética similar á dos inhibidores irreversibles que forman enlaces covalentes. Nestes casos, algúns destes inhibidores únense rapidamente ao encima formando un complexo EI de baixa afinidade, que despois experimenta unha reacción máis lenta cara a un complexo EI* moi fortemente unido (ver a imaxe arriba). Este comportamento cinético denomínase unión lenta.[18] Este lento reaxuste posterior normalmente implica un cambio conformacional cando o encima se prega arredor da molécula inhibidora. Son exemplos de inhibidores de unión lenta algún fármaco importante como o metotrexato,[19] o alopurinol [20] e a forma activada do aciclovir.[21]
Exemplos de inhibidores irreversibles
editarO diisopropilfluorofosfato (DFP) púxose como un exemplo de inhibidor irreversible da protease no apartado "Inhibidores irreversibles" arriba á dereita. O encima hidroliza o enlace entre o fósforo e o flúor, pero o residuo de fosfato mantense unido a unha serina no centro activo, inactivándoo.[22] Ademais, o DFP tamén reacciona co centro activo da acetilcolinesterase na sinapse das neuronas, o que o converte nunha potente neurotoxina, cunha dose letal a partir de cantidades inferiores a 100 mg.[23]
A chamada inhibición suicida é un tipo común de inhibición irreversible na que o encima converte ao inhibidor nunha substancia reactiva no seu centro activo. Un exemplo disto é o inhibidor de biosintetizadores de poliaminas α-difluorometilornitina ou DFMO, que é un análogo do aminoácido ornitina, e utilízase para tratar a tripanosomíase africana (enfermidade do sono). A ornitina descarboxilase pode catalizar a descarboxilación do DFMO substituíndo a ornitina, como se mostra na figura do apartado anterior. Porén, esta reacción de descarboxilación vai seguida da eliminación do átomo de flúor, o que converte a este intermediario nunha imina, unha especie altamente electrofílica. Esta forma reactiva do DFMO reacciona posteriormente cun residuo de cisteína ou lisina no centro activo para inactivar o encima irreversiblemente.[17]
Como a inhibición irreversible implica con frecuencia a formación inicial dun complexo EI non covalente, ás veces é posible que un inhibidor poida unirse a un encima de diversas formas. Como exemplo serve o caso do encima tripanotión redutase do protozoo e parasito humano Trypanosoma cruzi, no que dúas moléculas dun inhibidor chamado mostaza quinacrina, presentan a capacidade de unirse ao seu centro activo. A molécula superior únese de forma reversible, pero a de abaixo únese de forma covalente ao reaccionar cun residuo de aminoácido a través do seu grupo de mostaza nitroxenada.[24]
Descubrimento e deseño de inhibidores
editarA investigación e desenvolvemento de novos fármacos é un longo proceso no cal o primeiro paso case sempre é o descubrimento dun novo inhibidor encimático. No pasado, a única forma de descubrir estes novos inhibidores era por medio do sistema de proba e erro: probando un elevado número de compostos contra o encima ao cal se quere inhibir e esperando atopar algún que sexa efectivo. Esta aproximación, aínda que pouco refinada, segue obtendo actualmente un grande éxito grazas a novos sistemas de varrido, como a química combinatoria, que rapidamente producen un gran número de compostos novidosos, e á tecnoloxía baseada na investigación de procesamento de alto-rendemento, por medio da cal se poden revisar rapidamente estas enormes bibliotecas químicas de inhibidores útiles.[25]
Máis recentemente, comezouse a aplicar un novo tipo de investigación alternativa: o deseño racional de fármacos que usa a estrutura tridimensional do sitio activo dun encima para predicir que moléculas poderían ser inhibidores.[26] Unha vez realizada a predición, próbanse e selecciónase o mellor. Este novo inhibidor úsase despois para tratar de obter unha estrutura do encima nun complexo encima-substrato para mostrar como se une a molécula ao sitio activo. Esta estrutura é despois inspeccionada e lévanse a cabo certos cambios na estrutura do inhibidor coa fin de optimizar a unión co encima. Este ciclo de proba e optimización repítese de forma sucesiva ata obter un inhibidor dabondo potente, é dicir, cando se atingue un valor da constante de disociación que estea arredor de <10−9 M.[27]
Aplicacións dos inhibidores
editarOs inhibidores encimáticos poden encontrarse na natureza, mais tamén son deseñados e producidos como parte da farmacoloxía e a bioquímica. Os velenos naturais son a miúdo inhibidores encimáticos que evolucionaron para defender a unha planta ou animal contra os seus depredadores. Estas toxinas naturais inclúen algúns dos compostos máis velenosos coñecidos ata hoxe. Os inhibidores artificiais son con frecuencia usados como medicamentos, pero tamén existen algúns utilizados como insecticidas (como o malatión), herbicidas (como o glifosato) ou desinfectantes, (como o triclosán).
Fármacos
editar|
|
|
|
|
|
Os inhibidores encimáticos utilízanse principalmente como fármacos no tratamento de diversas enfermidades. Moitos destes inhibidores poden actuar sobre encimas humanos e así corrixir determinadas patoloxías. Porén, non todos os fármacos inhibitorios son inhibidores encimáticos. Algúns deles, tales como os fármacos anti-epilépticos, alteran a actividade encimática de forma indirecta, aumentando ou diminuíndo a síntese de dito encima. Estes efectos denomínanse indución e inhibición encimática e consisten en alteracións no patrón de expresión xénica, o cal non está relacionado co tipo de inhibición encimática discutido aquí. Outras drogas interaccionan con outras dianas celulares que non son encimas, como os canles iónicas ou os receptores de membrana.[28][29]
Un exemplo de inhibidor encimático terapéutico é o sildenafilo (Viagra), utilizado como tratamento da disfunción eréctil. Este composto é un potente inhibidor da fosfodiesterase tipo 5 específica de GMPc, un encima que degrada unha molécula de sinalización celular, o GMPc.[30] Esta molécula de sinalización é a responsable da relaxación do músculo liso e permite o fluxo de sangue cara ao interior dos corpos cavernosos do pene, o cal causa a erección. O sildenafilo inhibe a actividade do encima que degrada o sinal, o GMPc, uníndose ao mesmo sitio ca este debido á súa semellanza estrutural. Isto permite que o GMPc non sexa degradado e poida permanecer activo durante períodos máis longos de tempo.[30]
Outro exemplo de semellanza estrutural entre inhibidores e substratos encimáticos é que o que se mostra na figura da dereita, entre o metotrexato, un fármaco, e o ácido fólico, un coencima. O ácido fólico é a forma oxidada do substrato da dihidrofolato redutase, un encima implicado na biosíntese de timidina, purinas e aminoácidos. O metotrexato é un potente inhibidor deste encima que, ao estar relacionado coa síntese de nucleótidos, presenta unha toxicidade específica para as células cunha rápida taxa de crecemento. O metotrexato utilízase a miúdo como fármaco anticanceríxeno en quimioterapia.[31]
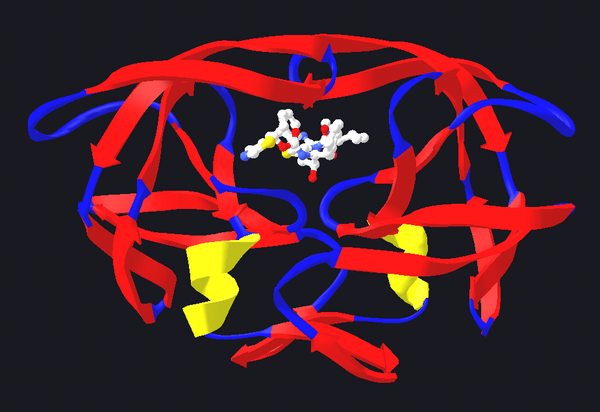
Outro tipo de inhibidores encimáticos utilízanse coa fin de inhibir os encimas necesarios para a supervivencia de patóxenos. Por exemplo, as bacterias presentan unha grosa parede celular composta principalmente por un polímero denominado peptidoglicano. Certos antibióticos como a penicilina e a vancomicina inhiben ao encima responsable da produción e o entrecruzamento das fibras de peptidoglicano,[32] o cal dá lugar a unha perda de forza da parede celular e á lise da célula, que xa non pode resistir a elevada presión osmótica. Na figura pódese apreciar unha molécula de penicilina unida á súa diana, a transpeptidase da bacteria Streptomyces R61 (a proteína móstrase no modelo de fitas e a penicilina nun modelo de esferas e barras). Tamén se utilizan outro tipo de toxicidades selectivas por medio de antibióticos que aproveitan as diferenzas presentes na estrutura dos ribosomas ou na síntese de ácidos graxos. O deseño de fármacos está moi facilitado nestes casos nos que o encima diana é esencial para a supervivencia do patóxeno e non está presente ou é moi diferente en humanos.[32]
Control metabólico
editarOs inhibidores encimáticos son tamén importantes no control metabólico. Moitas das rutas metabólicas que teñen lugar na célula son inhibidas por metabolitos que controlan a actividade encimática por procesos de regulación alostérica ou inhibición por substrato. Como exemplo podemos mencionar a regulación alostérica da glicólise. Esta ruta catabólica consome glicosa e produce ATP, NADH e piruvato. Unha das fases chave na regulación da glicólise é a reacción catalizada pola fosfofructoquinase-1 (PFK1). Cando os niveis de ATP aumentan, o ATP únese a un sitio alostérico da PFK1, co que se reduce a actividade do encima, a glicólise inhíbese e os niveis de ATP redúcense de novo. Este control por retroalimentación negativa (feedback negativo) axuda a manter os niveis de ATP constantes na célula. Porén, as rutas metabólicas non están unicamente reguladas por medio da inhibición; a activación encimática é igualmente importante. O encima PFK1 presenta activadores alostéricos, como son a frutosa 2,6-bifosfato e o ADP.[33]
A inhibición encimática fisiolóxica tamén pode producirse por inhibidores proteicos específicos. Este mecanismo pódese observar no páncreas, órgano onde se sintetizan multitude de precursores de encimas dixestivos denominados cimóxenos. Moitos deles son activados pola tripsina, unha protease, polo que é moi importante inhibir a actividade da tripsina no páncreas para previr fenómenos de autodixestión. Un dos mecanismos para manter a tripsina inactiva é a produción dun potente e específico inhibidor de tripsina no páncreas. Este inhibidor únese cunha alta afinidade á tripsina, previndo así a súa actividade.[34] Aínda que o inhibidor de tripsina é unha proteína, non é hidrolizado pola propia tripsina, xa que elimina as moléculas de auga do seu centro activo e desestabiliza o estado de transición.[35] Outros exemplos de inhibidores encimáticos fisiolóxicos son o inhibidor barstar, que ten a función de inhibir a actividade dunha ribonuclease bacteriana denominada barnase,[36] e os inhibidores das proteín-fosfatases.[37]
Inhibidores artificiais
editarInhibidores da acetilcolinesterase
editarA acetilcolinesterase (AChE) é un encima que se encontra nos animais, desde os insectos ata os humanos. É esencial na actividade que desenvolven as neuronas, xa que a súa función consiste en romper o neurotransmisor acetilcolina nos seus constituíntes, acetato e colina.[38][39] É un caso único entre os neurotransmisores, xa que a maioría deles, como a serotonina, a dopamina ou a noradrenalina, son reabsorbidos na fenda sináptica. Existe un amplo número de inhibidores da AChE que son utilizados tanto no campo da medicina coma no campo da agricultura. Algúns destes inhibidores son reversibles, como o edrofonio, a fisostigmina, e a neostigmina,[40] os cales son utilizados no tratamento da miastenia gravis e na aplicación de anestesia. Os pesticidas do tipo carbamato son outro exemplo de inhibidores reversibles da AChE. Como exemplo de inhibidores irreversibles da AChE están os insecticidas organofosforados, como o malathion, o paratión e o clorpirifós.[40]
Herbicidas
editarO glifosato é un herbicida non selectivo de amplo espectro, desenvolvido para a eliminación de herbas e arbustos, en especial daqueles de natureza perenne. É absorbido polas follas e non polas raíces. Pódese aplicar ás follas, inxectarse a troncos e talos, ou regado sobre tocos de árbores como herbicida forestal. A aplicación de glifosato mata as plantas debido a que suprime a súa capacidade de xerar aminoácidos aromáticos. O seu modo de acción baséase na inhibición do encima 5-enolpiruvil-siquimato-3-fosfato sintetase (EPSPS), encima responsable da síntese dos aminoácidos aromáticos fenilalanina, tirosina e triptófano. Esta ruta bioquímica non está presente en animais, polo que non se ven afectados.[41] Outros herbicidas, como as sulfonilureas inhiben o encima acetolactato sintase.
Desinfectantes
editarO triclosán é un potente axente antibacteriano e funxicida, actuando como biocida sobre diversas dianas no citoplasma e a membrana plasmática.[42] Porén, a baixas concentracións, o triclosán actúa como axente bacteriostático principalmente inhibindo a síntese de ácidos graxos. O triclosán únese á proteína transportadora enoíl‐acil redutase (ENR), codificada polo xene FabI. Esta unión aumenta a afinidade do encima polo NAD+, o cal orixina a formación dun complexo ternario estable de ENR-NAD+-triclosán, que non pode participar na síntese de ácidos graxos (moléculas imprescindibles na construción e mantemento das membranas celulares). O ser humano non posúe o encima ENR, de modo que non se ve afectado polo triclosán.[42]
Velenos naturais
editarTanto algúns animais como algunhas plantas poden sintetizar unha ampla gama de substancias velenosas de diversa orixe: metabolitos secundarios, péptidos e proteínas, que poden actuar como inhibidores encimáticos. As toxinas naturais adoitan ser pequenas moléculas orgánicas con tanta diversidade que probablemente existan inhibidores para a maioría dos procesos metabólicos.[43] Dita inhibición pode producirse a diferentes niveis na célula: inhibición de receptores de membrana, de canles iónicas ou de proteínas estruturais. Por exemplo, o paclitaxel (taxol), unha molécula orgánica producida polo teixo (Taxus), únese cunha elevada afinidade aos dímeros de tubulina, impedindo así o proceso de polimerización dos microtúbulos, fundamental, entre outras cousas, na división celular.[44]
Moitos destes velenos naturais actúan como neurotoxinas que poden causar parálises e levar á morte, e poden utilizarse como estratexia defensiva fronte aos depredadores ou como sistema de caza na captura de presas. Algúns destes inhibidores naturais, malia as súas características tóxicas, son moi valorados polos seus potenciais usos terapéuticos cando son administrados en doses adecuadas.[45] Un exemplo de neurotoxina son os glicoalcaloides, obtidos a partir das plantas pertencentes á familia Solanaceae (entre as que se inclúen a pataca, o tomate e a berenxena), que son inhibidores da acetilcolinesterase. A inhibición deste encima causa un aumento descontrolado dos niveis do neurotransmisor acetilcolina, o que comporta parálise muscular e morte. A neurotoxicidade tamén pode ser o resultado da inhibición de receptores, como no caso da atropina, obtida da planta da especie Atropa belladonna, que funciona como un antagonista competitivo dos receptores muscarínicos de acetilcolina.[46]
Aínda que moitas das toxinas naturais son metabolitos secundarios, algunhas son péptidos ou proteínas. Un exemplo é a toxina peptídica alfa-amanitina, encontrada nos fungos da especie Amanita phalloides, que é un potente inhibidor encimático que impide a actividade da ARN polimerase II na transcrición do ADN.[47] Outra toxina peptídica é a microcistina, encontrada en certas algas, que pode inhibir certas proteín-fosfatases.[48] Esta toxina pode contaminar as reservas de auga se as algas que a producen atinguen determinados niveis de concentración. Demostrouse o seu alto potencial carcinóxeno e a súa capacidade de causar hemorraxia hepática aguda e morte tras unha exposición a altas doses.[49]
As proteínas tamén poden chegar a ser velenos naturais, como é o caso do inhibidor de tripsina (discutido anteriormente) encontrado nalgúns legumes. Menos comúns son os encimas tóxicos. Este tipo de encimas actúan como inhibidores irreversibles de dianas encimáticas (o seu substrato é outro encima), modificándoas quimicamente. Por exemplo, a ricina, unha proteína tóxica encimática estremadamente potente, que se encontra na planta Ricinus communis. Este encima é unha glicosidase que inactiva os ribosomas. Como a ricina é un inhibidor catalítico irreversible, isto permite que tan só unha molécula de ricina poida matar unha célula.[50]
Notas
editar- ↑ Shapiro R, Vallee BL. Interaction of human placental ribonuclease with placental ribonuclease inhibitor. Biochemistry. 1991 Feb 26;30(8):2246–55. (en inglés) PMID 1998683
- ↑ 2,0 2,1 2,2 2,3 Berg J., Tymoczko J. and Stryer L. (2002) Biochemistry. W. H. Freeman and Company (en inglés) ISBN 0-7167-4955-6
- ↑ 3,0 3,1 3,2 3,3 3,4 3,5 3,6 * Irwin H. Segel, Enzyme Kinetics : Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. Wiley–Interscience; New edition (1993), (en inglés) ISBN 0-471-30309-7
- ↑ Holdgate GA. Making cool drugs hot: isothermal titration calorimetry as a tool to study binding energetics. Biotechniques. 2001 Jul;31(1):164–6 (en inglés) PMID 11464510
- ↑ 5,0 5,1 Leatherbarrow RJ. Using linear and non-linear regression to fit biochemical data. Trends Biochem Sci. 1990 Dec;15(12):455–8. (en inglés) PMID 2077683
- ↑ Tseng SJ, Hsu JP. A comparison of the parameter estimating procedures for the Michaelis–Menten model. J Theor Biol. 1990 Aug 23;145(4):457–64. (en inglés) PMID 2246896
- ↑ 7,0 7,1 Dixon, M. Webb, E.C., Thorne, C.J.R. and Tipton K.F., Enzymes (3rd edition) Longman, London (1979) ver p. 126 (en inglés)
- ↑ Hsu JT, Wang HC, Chen GW, Shih SR. Antiviral drug discovery targeting to viral proteases. Curr Pharm Des. 2006; 12(11):1301–14. (en inglés) PMID 16611117
- ↑ Lew W, Chen X, Kim CU (2000). "Discovery and development of GS 4104 (oseltamivir): an orally active influenza neuraminidase inhibitor". Curr. Med. Chem. (en inglés) 7 (6): 663–72. PMID 10702632.
- ↑ Fischer PM (2003). "The design, synthesis and application of stereochemical and directional peptide isomers: a critical review". Curr. Protein Pept. Sci. (en inglés) 4 (5): 339–56. PMID 14529528.
- ↑ Bogoyevitch MA, Barr RK, Ketterman AJ. Peptide inhibitors of protein kinases—discovery, characterisation and use. Biochim Biophys Acta. 2005 Dec 30;1754(1-2):79–99. (En inglés) PMID 16182621
- ↑ Lundblad R. L. Chemical Reagents for Protein Modification CRC Press Inc (2004). (En inglés) ISBN 0-8493-1983-8
- ↑ N. Price, B. Hames, D. Rickwood (Ed.) Proteins LabFax Academic Press (1996). (En inglés) ISBN 0-12-564710-7
- ↑ 14,0 14,1 Adam GC, Cravatt BF, Sorensen EJ. (2001) Profiling the specific reactivity of the proteome with non-directed activity-based probes. Chem. Biol. 8(1):81-95. (En inglés)
- ↑ 15,0 15,1 Maurer T, Fung HL. Comparison of Methods for Analyzing Kinetic Data From Mechanism-Based Enzyme Inactivation: Application to Nitric Oxide Synthase. AAPS PharmSci. (2000) 2(1)E8. (En inglés) PMID 11741224
- ↑ Loo JA, DeJohn DE, Du P, Stevenson TI, Ogorzalek Loo RR (1999). "Application of mass spectrometry for target identification and characterization". Med Res Rev (en inglés) 19 (4): 307–19. PMID 10398927.
- ↑ 17,0 17,1 Poulin R, Lu L, Ackermann B, Bey P, Pegg AE. Mechanism of the irreversible inactivation of mouse ornithine decarboxylase by alpha-difluoromethylornithine. Characterization of sequences at the inhibitor and coenzyme binding sites. Arquivado 24 de xaneiro de 2009 en Wayback Machine. J Biol Chem. 1992 Jan 5;267(1):150–8. (En inglés) PMID 1730582
- ↑ Szedlacsek, S.E. and Duggleby, R.G. Kinetics of slow and tight-binding inhibitors. Meth. Enzymol., (1995) 249: 144–180. (En inglés) PMID 7791610
- ↑ Stone SR, Morrison JF. Mechanism of inhibition of dihydrofolate reductases from bacterial and vertebrate sources by various classes of folate analogues. Biochim Biophys Acta. 1986 Feb 14;869(3):275–85. (En inglés) PMID 3511964
- ↑ Hille R, Massey V. Tight binding inhibitors of xanthine oxidase. Pharmacol Ther. 1981;14(2):249–63. (En inglés) PMID 4322209
- ↑ Reardon JE. Herpes simplex virus type 1 and human DNA polymerase interactions with 2'-deoxyguanosine 5'-triphosphate analogues. Kinetics of incorporation into DNA and induction of inhibition. Arquivado 24 de xaneiro de 2009 en Wayback Machine. J Biol Chem. 1989 Nov 15;264(32):19039–44. (En inglés) PMID 2553730
- ↑ J. A. Cohen , R. A. Oosterbaan and F. Berends Organophosphorus compounds Meth. Enzymol. (1967) 11, 686. (En inglés)
- ↑ Brenner, G. M. (2000): Pharmacology. Philadelphia, PA: W.B. Saunders Company. (En inglés) ISBN 0-7216-7757-6
- ↑ Saravanamuthu A, Vickers TJ, Bond CS, Peterson MR, Hunter WN, Fairlamb AH. Two interacting binding sites for quinacrine derivatives in the active site of trypanothione reductase: a template for drug design. Arquivado 12 de maio de 2009 en Wayback Machine. J Biol Chem. 2004 Jul 9;279(28):29493–500. (En inglés) PMID 15102853
- ↑ Koppitz M, Eis K (2006). "Automated medicinal chemistry". Drug Discov. Today (en inglés) 11 (11-12): 561–8. PMID 16713909. doi:10.1016/j.drudis.2006.04.005.
- ↑ Scapin G (2006). "Structural biology and drug discovery". Curr. Pharm. Des. (en inglés) 12 (17): 2087–97. PMID 16796557. doi:10.2174/138161206777585201.
- ↑ Hunter WN. Rational drug design: a multidisciplinary approach. Mol Med Today. 1995 Apr;1(1):31, 34. (En inglés) PMID 9415135
- ↑ Patsalos PN. Antiepileptic drug pharmacogenetics. Ther Drug Monit. 2000 Feb;22(1):127-30. (En inglés) PMID 10688275
- ↑ Armijo JA, de las Cuevas I, Adín J. Ion channels and epilepsy. Rev Neurol. 2000 Jun;30 Suppl 1:S25-41. (En castelán e inglés) PMID 10904966
- ↑ 30,0 30,1 Maggi M, Filippi S, Ledda F, Magini A, Forti G. Erectile dysfunction: from biochemical pharmacology to advances in medical therapy. Arquivado 28 de setembro de 2007 en Wayback Machine. Eur J Endocrinol. 2000 Aug;143(2):143–54. (En inglés) PMID 10913932
- ↑ McGuire JJ. Anticancer antifolates: current status and future directions. Curr Pharm Des. 2003;9(31):2593–613. (En inglés) PMID 14529544
- ↑ 32,0 32,1 Katz AH, Caufield CE. Structure-based design approaches to cell wall biosynthesis inhibitors. Curr Pharm Des. 2003;9(11):857–66. (En inglés) PMID 12678870
- ↑ Okar DA, Lange AJ. Fructose-2,6-bisphosphate and control of carbohydrate metabolism in eukaryotes. Biofactors. 1999;10(1):1–14. (En inglés)
- ↑ Nicholas Price, Lewis Stevens, Fundamentals of Enzymology, Oxford University Press, (1999). (En inglés) ISBN 0-19-850229-X
- ↑ Smyth TP. Substrate variants versus transition state analogues as noncovalent reversible enzyme inhibitors. Bioorg Med Chem. 2004 Aug 1;12(15):4081–8. (En inglés) PMID 15246086
- ↑ Hartley RW. Barnase and barstar: two small proteins to fold and fit together. Trends Biochem Sci. 1989 Nov;14(11):450–4. (En inglés) PMID 2696173
- ↑ Oliver CJ, Shenolikar S. Physiologic importance of protein phosphatase inhibitors. Front Biosci. 1998 Sep 1;3:D961–72. (En inglés) PMID 9727084
- ↑ Pauling, Lynus (1946). "Molecular Architecture and Biological Reactions" (PDF). Chemical Engineering News (en inglés) 24: 1375.
- ↑ Fersht, Alan (1985). W.H. Freeman, ed. Enzyme structure and mechanism (en inglés). San Francisco. p. 14. ISBN 0-7167-1614-3.
- ↑ 40,0 40,1 Rang, H. P. (2003). Pharmacology (en inglés). Edinburgh: Churchill Livingstone. pp. 156. ISBN 0-443-07145-4.
- ↑ Steinrücken HC, Amrhein N (1980). "The herbicide glyphosate is a potent inhibitor of 5-enolpyruvyl-shikimic acid-3-phosphate synthase". Biochem Biophys Res Commun 94: 1207–1212. PMID 7396959. doi:10.1016/0006-291X(80)90547-1..
- ↑ 42,0 42,1 Russell AD (2004). "Whither triclosan?". J. Antimicrob. Chemother. 53 (5): 693–5. PMID 15073159. doi:10.1093/jac/dkh171.
- ↑ Tan G, Gyllenhaal C, Soejarto DD. Biodiversity as a source of anticancer drugs. Curr Drug Targets. 2006 Mar;7(3):265-77. (En inglés) PMID 16515527
- ↑ Abal M, Andreu JM, Barasoain I. Taxanes: microtubule and centrosome targets, and cell cycle dependent mechanisms of action. Curr Cancer Drug Targets. 2003 Jun;3(3):193–203. (En inglés) PMID 12769688
- ↑ Hostettmann K, Borloz A, Urbain A, Marston A, Natural Product Inhibitors of Acetylcholinesterase Current Organic Chemistry, 2006 May;10(8):825–47. (En inglés)
- ↑ DeFrates LJ, Hoehns JD, Sakornbut EL, Glascock DG, Tew AR. Antimuscarinic intoxication resulting from the ingestion of moonflower seeds. Ann Pharmacother. 2005 Jan;39(1):173-6. (En inglés) PMID 15572604
- ↑ Vetter J. Toxins of Amanita phalloides. Arquivado 12 de maio de 2009 en Wayback Machine. Toxicon. 1998 Jan;36(1):13–24. (En inglés) PMID 9604278
- ↑ Holmes CF, Maynes JT, Perreault KR, Dawson JF, James MN. Molecular enzymology underlying regulation of protein phosphatase-1 by natural toxins. Curr Med Chem. 2002 Nov;9(22):1981–9. (En inglés) PMID 12369866
- ↑ Bischoff K. The toxicology of microcystin-LR: occurrence, toxicokinetics, toxicodynamics, diagnosis and treatment. Vet Hum Toxicol. 2001 Oct;43(5):294-7. (En inglés) PMID 11577938
- ↑ Hartley MR, Lord JM. Cytotoxic ribosome-inactivating lectins from plants. Arquivado 12 de maio de 2009 en Wayback Machine. Biochim Biophys Acta. 2004 Sep 1;1701(1–2):1–14. (En inglés) PMID 15450171
Véxase tamén
editarOutros artigos
editarLigazóns externas
editar- Tutorial sobre inhibición encimática
- Simboloxía e terminoloxía na cinética encimática
- Base de datos de fármacos e inhibidores encimáticos (PubChem del NCBI
- BRENDA: base de datos de encimas e os seus correspondentes inhibidores coñecidos Arquivado 11 de decembro de 2008 en Wayback Machine.
- Encimas, Cinética e Diagnóstico. Aplicacións médicas dos inhibidores encimáticos Arquivado 09 de xullo de 2011 en Wayback Machine.
- BindingDB: base de datos pública que recolle os valores de afinidades de unión proteína-ligando
- Exercicios animados de inhibición encimática: tutoriais e preguntas